Vagal Paragangliomas
Mark S. Persky
INTRODUCTION
Paragangliomas represent highly vascular neoplasms embryologically arising from the paraganglia of neural crest origin and most commonly occur in the head and neck region. Paraganglia are part of the diffuse neuroendocrine system, previously known as the amine precursor decarboxylate system, and have the potential to secrete neuropeptides and catecholamines. Vagal paraganglia are normal structures that are intimately associated with the perineurium of the vagus nerve. While they may occur anywhere along the course of the vagus nerve, they are most often located within or just below the nodose (inferior) ganglion, which is just inferior to the skull base at the jugular foramen, and less often within the middle and jugular (superior) ganglion. Its location in this area predisposes vagal paragangliomas to early involvement of the other lower cranial nerves, including the glossopharyngeal, accessory, and hypoglossal nerves, and dysfunction of these multiple lower cranial nerves may be apparent with larger or long-standing tumors. Multicentric paragangliomas are common in familial cases (78% to 87%) but also occur in 10% of sporadic tumors, and appropriate studies should be performed to identify the presence of additional paragangliomas. Malignant vagal paragangliomas are uncommon, and their diagnosis can only be confirmed by the presence of metastatic tumor, usually within regional lymph nodes. There are no strict histologic criteria within the primary tumor that can differentiate between benign and malignant paragangliomas. Paragangliomas are highly vascular, and they characteristically demonstrate early neural involvement in addition to skull base and potential intracranial extension. These factors all contribute to the challenging nature of effectively treating these tumors. Traditionally, surgery has been the preferred method of treatment, especially with the evolution of more sophisticated cervical and skull base approaches. Postoperative cranial nerve dysfunction may be anticipated in patients with larger tumors and skull base involvement; therefore, a focus on rehabilitation efforts is necessary.
Approximately 1% to 3% of paragangliomas secrete catecholamines. A fivefold increase in catecholamines is sufficient to produce symptoms. Symptoms consistent with a functioning tumor are tachycardia, excessive sweating, weight loss, and hypertension. Secreting paragangliomas account for approximately 2% of all instances of secondary hypertension. Twenty-four-hour urine collection in these patients will show elevated levels of the catecholamine metabolites, metanephrine and vanillylmandelic acid. Serum catecholamines will show elevated levels of norepinephrine in functioning extra-adrenal paragangliomas. Elevated serum epinephrine is indicative of a concurrent pheochromocytoma. Paraganglioma familial syndrome subtypes 1 and 4 (PGL1 and PGL4) are associated with a higher incidence of concurrent pheochromocytoma. Conversely, multiple endocrine neoplasia type IIA (pheochromocytoma, medullary thyroid carcinoma, and parathyroid hyperplasia) and type IIB (pheochromocytoma, medullary thyroid carcinoma, parathyroid hyperplasia, and mucosal neuroma) are associated with head and neck paragangliomas.
HISTORY
Vagal paragangliomas are slow growing with a female:male preponderance of 2:1 to 3:1 and a mean duration of symptoms of 2 to 3 years before presentation. The most common symptom is a slowly growing, asymptomatic mass in the superior aspect of the neck. As the tumor enlarges, it encroaches upon the lower cranial nerves and the adjacent sympathetic chain. Approximately 33% to 50% of patients have cranial nerve neuropathy at pre-sentation involving, in decreasing frequency, the vagal, hypoglossal, spinal accessory, and sympathetic plexus nerves. Signs and symptoms include unilateral vocal cord paralysis, hoarseness, dysphagia, nasal regurgitation, atrophy of the hemitongue, shoulder weakness, and Horner syndrome. The initial presence of vocal cord paralysis with or without hoarseness helps to differentiate vagal paragangliomas from carotid body tumors.
Vagal paragangliomas account for up to 5% of all head and neck paragangliomas. While they arise most commonly from the nodose (inferior) ganglion, vagal paragangliomas can originate from the middle and superior ganglion and less frequently anywhere along the course of the vagus nerve. Compared to the discrete carotid body, vagal paraganglia are distributed more diffusely within the nerve or perineurium. Vagus nerve fibers fan out or “splay over” the surface of the vagal paraganglioma or, early in their development, enter the substance of the tumor, and therefore, preservation of the vagus nerve is usually not possible with complete tumor resection.
Vagal paragangliomas are ovoid- or spindle-shaped tumors and most commonly present as an asymptomatic mass in the superior aspect of the neck, typically more cephalad than carotid body tumors. Since most vagal paragangliomas originate at the inferior (nodose) ganglion, the tumor tends to spread inferiorly into the poststyloid parapharyngeal area. Extension superiorly toward the skull base in the area of the jugular foramen results in early involvement of the internal jugular vein, adjacent cranial nerves (IX, XI, XII) (Fig. 12.1), and internal carotid artery, manifesting the typical anterior/medial displacement of this artery (Fig. 12.2). The vagus nerve is located in the poststyloid compartment of the parapharyngeal space (Fig. 12.3).
PHYSICAL EXAMINATION
Vagal paragangliomas present as slowly growing, firm masses in level II of the neck. Although these represent vascular tumors, they generally do not present as pulsatile masses although there may be associated bruits. The mobility of the tumor to palpation reveals that it is fixed in a vertical direction due to its intimate involvement with the vagus nerve. Medial parapharyngeal extension may cause a bulge of the lateral wall of the oropharynx and medial displacement of the tonsil. With enlargement of the tumor, both the vagus and adjacent cranial nerves are involved. The vagus nerve is affected most commonly, followed by the hypoglossal and spinal accessory nerves; therefore, ipsilateral vocal cord paresis/paralysis, weakness of the tongue, and atrophy of the sternocleidomastoid and trapezius muscles may be presenting findings. Involvement of the sympathetic plexus may cause Horner syndrome.
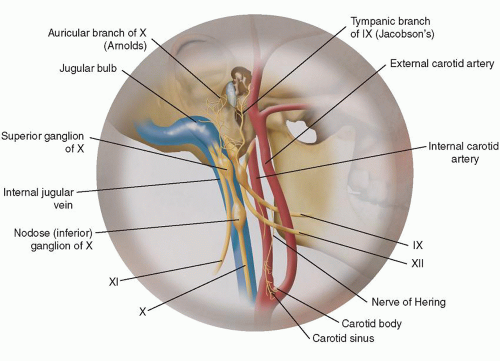 FIGURE 12.1 Illustration of the anatomy of the superior aspect of the vagus nerve with adjacent structures at risk for involvement with an enlarging vagal paraganglioma. |
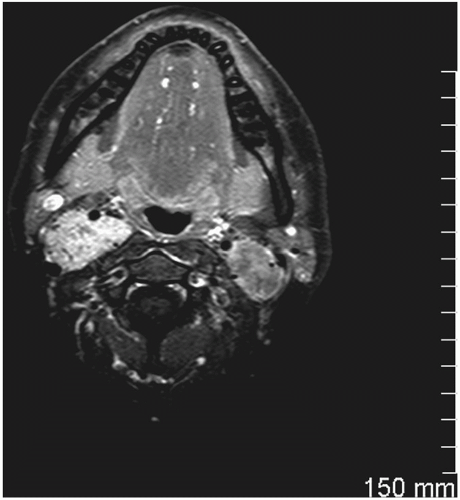 FIGURE 12.2 MRI of a patient with bilateral vagal paragangliomas and typical anterior-medial displacement of the internal carotid arteries. |
INDICATIONS
Surgery is indicated for patients presenting with paresis/paralysis of the vagus nerve assuming that advanced age, physical disabilities, and comorbidities are not contraindications. One should assume that total dysfunction of the vagus nerve will result and that adjacent cranial nerve dysfunction will possibly be a sequela. One must realize that paragangliomas are radiosensitive and that doses of 45 Gy delivered over 5 weeks can prevent tumor growth and cranial nerve dysfunction in over 90% of patients. Documentation of a malignant vagal paraganglioma by imaging studies and subsequent needle aspiration biopsy will require primary tumor resection, modified neck dissection, and adjuvant radiation therapy.
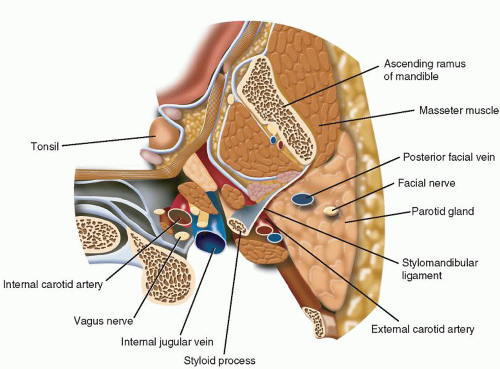 FIGURE 12.3 Anatomy of the parapharyngeal space indicating the position of the vagus nerve in the poststyloid compartment. |



