Uveitis
12.1 Anterior Uveitis (Iritis/Iridocyclitis)
Symptoms
Acute: Pain, redness, photophobia, consensual photophobia (pain in the affected eye when a light is shone in the fellow eye), excessive tearing, decreased vision.
Chronic: Decreased vision (from cataract, vitreous debris, cystoid macular edema (CME), or epiretinal membrane [ERM]) and floaters. May have periods of exacerbations and remissions with few acute symptoms (e.g., juvenile idiopathic arthritis [JIA]).
Signs
Critical. Cells and flare in the anterior chamber, ciliary flush, keratic precipitates (KP):
Fine KP (“stellate;” typically covers entire corneal endothelium): Herpes simplex or varicella zoster virus, cytomegalovirus (CMV), Fuchs heterochromic iridocyclitis (FHIC).
Small, nongranulomatous KP (NGKP): Human leukocyte antigen (HLA)-B27-associated, trauma, masquerade syndromes, JIA, Posner–Schlossman syndrome (glaucomatocyclitic crisis), drug-induced. Granulomatous uveitides such as sarcoidosis can present with NGKP; the reverse rarely occurs.
Granulomatous KP (large, greasy, “mutton-fat”; mostly on inferior cornea): Sarcoidosis, syphilis, tuberculosis (TB), JIA-associated, sympathetic ophthalmia, lens-induced, Vogt–Koyanagi–Harada (VKH) syndrome, and others.
Other. Low intraocular pressure (IOP) more commonly seen (secondary to ciliary body hyposecretion), elevated IOP can occur (e.g., herpetic, lens-induced, FHIC, Posner–Schlossman syndrome), fibrin (e.g., HLA-B27 or endophthalmitis), hypopyon (e.g., HLA-B27, Behçet disease, infectious endophthalmitis, rifabutin, tumor), iris nodules (e.g., sarcoidosis, syphilis, TB), iris atrophy (e.g., herpetic, oral fourth generation fluoroquinolones), iris heterochromia (e.g., FHIC), iris synechiae (especially HLA-B27, sarcoidosis), band keratopathy (especially JIA in younger patients, any chronic uveitis in older patients), uveitis in a “quiet eye” (consider JIA, FHIC, masquerade syndromes), CME (see Figure 12.1.1).
Differential Diagnosis
Intermediate or panuveitis with spillover into the anterior chamber: Mainly floaters and decreased vision, positive fundoscopic findings (SEE 12.3, POSTERIOR UVEITIS).
Traumatic iritis. SEE 3.5, TRAUMATIC IRITIS.
Posner–Schlossman syndrome: Recurrent episodes of very high IOP and minimal inflammation. SEE 9.8, GLAUCOMATOCYCLITIC CRISIS/POSNER–SCHLOSSMAN SYNDROME.
Drug-induced uveitis (e.g., rifabutin, cidofovir, sulfonamides, pamidronate, systemic fluoroquinolones [especially moxifloxacin], some chemotherapeutic drugs).
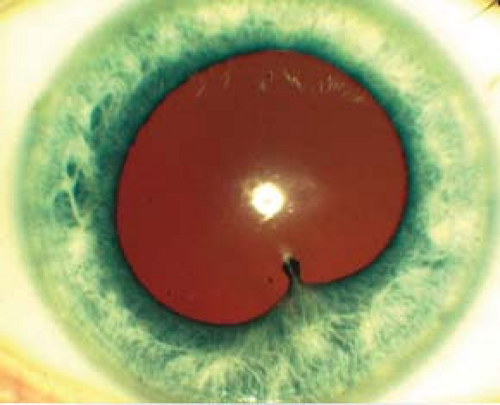
Figure 12.1.1 Anterior uveitis with posterior synechiae.
Sclerouveitis: Uveitis secondary to scleritis; typically presents with profound pain and tenderness to palpation. SEE 5.7, SCLERITIS.
CLARE (contact lens-associated red eye): Red eye, corneal edema, epithelial defects, iritis with or without hypopyon; hypoxic subepithelial or stromal infiltrates may be present.
Infectious keratouveitis: Corneal infiltrate is present. SEE 4.11, BACTERIAL KERATITIS.
Infectious endophthalmitis: History of recent surgery or penetrating trauma, pain, hypopyon, fibrinous anterior chamber reaction, vitritis, decreased vision, red eye; may have endogenous source with fever, elevated white blood cell count. SEE 12.13 TO 12.16, ENDOPHTHALMITIS SECTIONS.
Schwartz–Matsuo syndrome: Pigment released from a chronic retinal detachment clogs the trabecular meshwork, resulting in elevated IOP.
Tumor: Retinoblastoma in children, intraocular lymphoma in elderly, metastatic disease in all ages, and others.
Pseudouveitis from pigment dispersion syndrome. Other findings include Krukenburg spindle and iris transillumination defects. Pigment cells in the AC are smaller than white blood cells.
Etiology
Idiopathic (roughly half of all anterior uveitis has no identifiable cause or disease association).
HLA-B27-associated uveitis: Systemic associations include ankylosing spondylitis, reactive arthritis (Reiter syndrome), psoriatic arthritis, inflammatory bowel disease.
Lens-induced uveitis: Immune reaction to lens material, often secondary to incomplete cataract extraction, trauma with lens capsule damage, or hypermature cataract. SEE 9.12, LENS-RELATED GLAUCOMA.
Postoperative iritis: Anterior chamber inflammation following intraocular surgery. Endophthalmitis must be considered if severe inflammation and pain are present. SEE 12.14, CHRONIC POSTOPERATIVE UVEITIS.
Uveitis–Glaucoma–Hyphema (UGH) syndrome: Usually secondary to irritation from an intraocular lens (IOL) (particularly a closed-loop anterior chamber lens or single-piece lens in ciliary sulcus). SEE 9.16, POSTOPERATIVE GLAUCOMA.
Behçet disease: Young adults, acute shifting hypopyon, iritis, aphthous ulcers, genital ulcerations, erythema nodosum, retinal vasculitis (arteries and/or veins) and hemorrhages, may have recurrent episodes.
Lyme disease: May have history of a tick bite and rash. SEE 13.3, LYME DISEASE.
Anterior segment ischemia: Flare out of proportion to cellular reaction. Pain. Secondary to carotid insufficiency, tight scleral buckle, or previous extraocular muscle surgeries.
Tubulointerstitial nephritis and uveitis (TINU) syndrome: Rare, usually bilateral nongranulomatous uveitis in children and young adults, female predilection.
Other rare infectious etiologies of anterior uveitis: Mumps, influenza, adenovirus, measles, Chlamydia, Leptospirosis, Kawasaki disease, rickettsial disease, Chikungunya virus, and others.
Chronic
JIA: Usually occurs in young girls; may be painless and asymptomatic with minimal injection. Often bilateral. Iritis may precede the typical pauciarticular arthritis (four or fewer joints involved). Positive antinuclear antibody (ANA), negative rheumatoid factor, and increased erythrocyte sedimentation rate (ESR) most commonly seen. Associated with glaucoma, cataracts, band keratopathy, and CME. Uveitis may occur in polyarticular and rarely in systemic JIA (Still disease).
Chronic iridocyclitis of children: Usually occurs in young girls; is similar to JIA in signs and symptoms but lacks arthritis.
FHIC: Few symptoms, diffuse iris stromal atrophy often causing a lighter-colored iris with transillumination defects and blunting of the iris architecture. Gonioscopy may reveal fine vessels that cross the trabecular meshwork, typically without posterior synechiae. Fine KP over the entire corneal endothelium, mild anterior chamber reaction. Vitreous opacities, glaucoma, and cataracts are common, but macular edema and posterior synechiae are absent.
Sarcoidosis: More common in African Americans and Scandinavians. Usually bilateral; can have extensive posterior synechiae and conjunctival or iris nodules. SEE 12.6, SARCOIDOSIS.
Herpes simplex/varicella zoster: Diffuse KP, increased IOP, and iris atrophy. History of unilateral recurrent red eye, occasionally history of skin vesicles. Corneal scars associated with decreased corneal sensation may be present.
Syphilis: Anterior and intermediate uveitis most common. May have a maculopapular rash, iris roseola (vascular papules on the iris), and interstitial keratitis with ghost vessels in late stages. Inflammation of any ocular structure may occur. Placoid chorioretinitis is virtually pathognomonic. Neurosyphilis can cause meningismus. SEE 12.12, SYPHILIS.
Tuberculosis: Positive protein derivative of tuberculin (PPD) and/or interferon-gamma release assay (IGRA) (e.g., QuantiFERON-TB Gold), typical chest radiograph findings (helpful but not necessary for diagnosis; most TB uveitis occurs in patients without pulmonary TB), occasionally phlyctenular or interstitial keratitis, sometimes signs of posterior uveitis. SEE 12.3, POSTERIOR UVEITIS.
Others: Leprosy, brucellosis, etc.
Work-Up
Obtain a thorough history and review of systems (Tables 12.1.1 and 12.1.2). Specifically ask about fevers, chills, cough, shortness of breath, joint pain/swelling/stiffness, diarrhea, blood in urine/stool, skin rashes, and oral or genital ulcers.
 NOTE: Autoimmune diseases are less common in the very young and very old—consider masquerades.
NOTE: Autoimmune diseases are less common in the very young and very old—consider masquerades.
Complete ocular examination, including an IOP check, gonioscopy, and a dilated fundus examination. The vitreous should be evaluated for cells.
A laboratory work-up may be unnecessary in certain situations:
First episode of a mild, unilateral, nongranulomatous uveitis with a history and examination that is not suggestive of systemic disease.
Uveitis in the setting of known systemic disease such as sarcoidosis or the use of medicines known to cause uveitis (e.g., rifabutin).
Clinical findings are classic for a particular diagnosis (e.g., herpetic keratouveitis, FHIC, toxoplasmosis).
In all other situations requiring laboratory or diagnostic testing, performing a targeted work-up is recommended. If too many tests are ordered unnecessarily, a portion of them may come back false-positive and confuse the diagnosis. See Table 12.1.3. However, if a patient presents with bilateral, granulomatous, or recurrent uveitis without a suspected diagnosis, our practice is to at least evaluate for sarcoidosis, syphilis, and TB (in at-risk patients). Consider additional work-up as needed based on history and examination.
Rapid plasma reagin (RPR) or venereal disease research laboratory test (VDRL). Also need confirmatory test such as fluorescent treponemal antibody absorption (FTA-ABS) or treponemal-specific assay (e.g., microhemagglutination assay [MHA-TP]) given RPR and VDRL may be falsely negative.
Table 12.1.1 Epidemiology of Anterior Uveitis | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 12.1.2 Review of Systems | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 12.1.3 Suggested Diagnostic Work-Up for Anterior Uveitis | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
PPD and/or IGRA.
Chest radiograph or chest CT to rule out sarcoidosis and pulmonary tuberculosis.
Angiotensin-converting enzyme (ACE) ± lysozyme (questionable utility).
Lyme antibody (consider in endemic areas).
HLA-B27 (in acute unilateral or bilateral alternating anterior uveitis; especially if hypopyon present).
Anterior chamber paracentesis for polymerase chain reaction (PCR) testing for suspected herpes virus-associated anterior uveitis (CMV, herpes simplex, varicella zoster).
Treatment
Cycloplegic (e.g., cyclopentolate 1% t.i.d. for mild to moderate inflammation; atropine 1% b.i.d. to q.i.d. for severe inflammation).
Topical steroid (e.g., prednisolone acetate 1%) q1-6h, depending on severity of inflammation. Most cases of moderate to severe acute uveitis require q1–2h dosing initially. Difluprednate 0.05% may allow less frequent dosing than prednisolone acetate. Consider a loading dose (prednisolone acetate 1% one drop every minute for 5 minutes) or fluorometholone 0.1% ophthalmic ointment at night. If the anterior uveitis is severe, unilateral, and is not responding to topical steroids, then consider periocular repository steroids (e.g., 0.5 to 1.0 mL subtenon injection of triamcinolone 40 mg/mL). SEE APPENDIX 10, TECHNIQUE FOR RETROBULBAR/SUBTENON/SUBCONJUNCTIVAL INJECTIONS.
 NOTE: Periocular use of triamcinolone is off-label and must be discussed with patients. A trial of topical steroids at full strength for several weeks may help identify patients at risk of a significant IOP increase from steroids. Additionally, periocular depot steroids should be used with extreme caution in patients with scleritis because of possible scleral melting.
NOTE: Periocular use of triamcinolone is off-label and must be discussed with patients. A trial of topical steroids at full strength for several weeks may help identify patients at risk of a significant IOP increase from steroids. Additionally, periocular depot steroids should be used with extreme caution in patients with scleritis because of possible scleral melting.
If there is no improvement on maximal topical and repository steroids, or if the uveitis is bilateral and severe, consider systemic steroids, or immunosuppressive therapy. Consider referral to a uveitis specialist and rheumatologist.
 NOTE: Prior to initiating systemic steroids or periocular depot steroids, it is important to rule out infectious causes.
NOTE: Prior to initiating systemic steroids or periocular depot steroids, it is important to rule out infectious causes.
Treat secondary glaucoma with aqueous suppressants. Avoid pilocarpine. Glaucoma may result from:
Cellular blockage of the trabecular meshwork. SEE 9.7, INFLAMMATORY OPEN ANGLE GLAUCOMA.
Secondary angle closure from synechiae formation. SEE 9.4, ACUTE ANGLE CLOSURE GLAUCOMA.
Neovascularization of the iris and angle. SEE 9.14, NEOVASCULAR GLAUCOMA.
Steroid-response. SEE 9.9, STEROID-RESPONSE GLAUCOMA.
If an exact etiology for the anterior uveitis is determined, then additional ocular and/or systemic management may be indicated.
Ankylosing spondylitis: Often requires systemic anti-inflammatory agents (e.g., NSAIDs such as naproxen). Consider consulting rheumatology, physical therapy, and cardiology (increased incidence of cardiomegaly, conduction defects, and aortic insufficiency).
Inflammatory bowel disease (IBD): Often benefits from systemic steroids, sulfadiazine, or other immunosuppressive agents. Obtain a medical or gastrointestinal consult.
Reactive arthritis (previously known as Reiter syndrome): If urethritis is present, then the patient and sexual partners are treated for chlamydia (e.g., single dose azithromycin 1 g p.o.). Obtain medical and/or rheumatology or urology consult.
Psoriatic arthritis: Consider a rheumatology and/or dermatology consult.
Glaucomatocyclitic crisis: SEE 9.8, GLAUCOMATOCYCLITIC CRISIS/POSNER–SCHLOSSMAN SYNDROME.
Lens-induced uveitis: Usually requires removal of lens material. SEE 9.12, LENS-RELATED GLAUCOMA.
Herpetic uveitis: Herpes simplex typically requires topical or oral antivirals and steroid drops for non-epithelial corneal disease. Herpetic iridocyclitis benefits from topical steroids and systemic antiviral medications (e.g., acyclovir/valacyclovir); topical antivirals are ineffective for uveitis due to poor intraocular penetration. SEE 4.15, HERPES SIMPLEX VIRUS AND 4.16, HERPES ZOSTER OPHTHALMICUS/VARICELLA ZOSTER VIRUS.
UGH syndrome: SEE 9.16, POSTOPERATIVE GLAUCOMA.
Behçet disease: SEE 12.7, BEHÇET DISEASE.
Lyme disease: SEE 13.3, LYME DISEASE.
JIA: Steroid dosage is adjusted according to the degree of anterior chamber cells, not flare. Prolonged cycloplegic therapy may be required. Consult rheumatology or pediatrics as systemic steroid therapy or immunomodulatory therapy is often needed. Regular follow-up is essential as flares may be asymptomatic; recurrent or chronic disease can lead to irreversible damage and various sequelae including synechiae, glaucoma, CME, and cataract formation.
Chronic iridocyclitis of children: Same as JIA.
FHIC: Usually does not respond to or require steroids (a trial of steroids may be attempted, but they should be tapered quickly if there is no response); cycloplegics are not necessary.
Sarcoidosis: SEE 12.6, SARCOIDOSIS.
Syphilis: SEE 12.12, SYPHILIS.
Tuberculosis: Refer the patient to an internist, infectious disease specialist, or public health officer for consideration of systemic treatment. Patients with ocular TB frequently have no pulmonary disease but still require systemic four-drug antituberculous therapy. Concomitant oral steroids may be necessary.
Follow-Up
Every 1 to 7 days in the acute phase, depending on the severity; every 1 to 6 months when stable.
At each visit, the anterior chamber reaction and IOP should be evaluated.
A vitreous and fundus examination should be performed for all flare-ups, when vision is affected, or every 3 to 6 months. Macular edema is a frequent cause of decreased vision even after the uveitis is controlled; optical coherence tomography (OCT) can be very useful diagnostically.
If the anterior chamber reaction has resolved, then the steroid drops can be slowly tapered with intermittent examinations to ensure that the inflammation does not return during the taper (usually one drop per day every 3 to 7 days [e.g., q.i.d. for 1 week, then t.i.d. for 1 week, then b.i.d. for 1 week, etc.]). Steroids are usually discontinued following the taper when the anterior chamber does not have
any cellular reaction. Occasionally, long-term low-dose steroids every day or every other day are required to keep the inflammation from recurring. Punctal occlusion techniques may increase potency of drug and decrease systemic absorption. The cycloplegic agents also can be tapered off as the anterior chamber reaction improves and no new posterior synechiae are noted.
12.2 Intermediate Uveitis
Symptoms
Painless floaters and decreased vision. Minimal photophobia or external inflammation. Most often bilateral and classically affects patients age 15 to 40 years.
Signs
(See Figure 12.2.1.)
Critical. Vitreous cells and cellular aggregates floating predominantly in the inferior vitreous (snowballs). Younger patients may present with vitreous hemorrhage. White exudative material over the inferior ora serrata and pars plana (snowbank) is suggestive of pars planitis.
Other. Peripheral retinal vascular sheathing, peripheral neovascularization, mild anterior chamber inflammation, CME, posterior subcapsular cataract, band keratopathy, secondary glaucoma, ERM, and exudative retinal detachment. Posterior synechiae in pars planitis are uncommon and, if present, usually occur early in the course of disease.
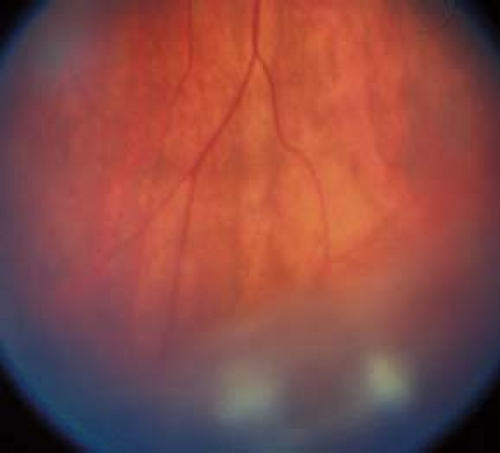 Figure 12.2.1 Pars planitis/intermediate uveitis with snowballs. |
Etiology
Work-Up
Complete ocular examination. Include gonioscopy to evaluate for possible neovascularization.
Appropriate work-up may include chest radiograph and/or chest CT, PPD and/or IGRA, ACE ± lysozyme, RPR or VDRL, and FTA-ABS or treponemal-specific assay.
Consider intravenous fluorescein angiography (IVFA) and/or OCT to document CME or retinal vasculitis.
Consider lab testing for Lyme disease, toxoplasmosis, cat-scratch disease in the appropriate clinical context. In older individuals, consider work-up for malignancy/lymphoma.
Consider magnetic resonance imaging (MRI) of the brain ± orbits with gadolinium to evaluate for demyelinating lesions if review of systems is positive for current or previous focal neurologic deficits. Refer to neurologist for multiple sclerosis work-up if necessary.
Treatment
Treat all vision-threatening complications (e.g., CME, vitritis) in symptomatic patients with active disease. Mild vitreous cell in the absence of symptoms or vision loss may be observed.
Topical prednisolone acetate 1% or difluprednate 0.05% q1–2h. Consider subtenon steroid (e.g., 0.5 to 1.0 mL injection of triamcinolone 40 mg/mL). May repeat the injections every 6 to 8 weeks until the vision and CME have stabilized. Slowly taper the frequency of injections. Subtenon steroid injections must be used with caution in patients with steroid-induced glaucoma. SEE APPENDIX 10, TECHNIQUE FOR RETROBULBAR/SUBTENON/SUBCONJUNCTIVAL INJECTIONS.
If minimal improvement after three subtenon steroid injections 1 to 2 months apart, consider systemic steroids (e.g., prednisone 40 to 60 mg p.o. daily for 4 to 6 weeks), tapering gradually according to the patient’s response. High-dose systemic steroid therapy should be no longer than 2 to 3 months, followed by a taper to no more than 5 to 10 mg/day. Other options include sustained-release steroid implants (e.g., dexamethasone 0.7 mg intravitreal implant; fluocinolone acetonide 0.19 or 0.59 mg intravitreal implant) and immunomodulatory therapy, often in conjunction with rheumatology.
 NOTE: In bilateral cases, systemic steroid therapy may be preferred to periocular injections. However, in children and adolescents, growth suppression (in addition to the usual complications of long-term systemic steroids) is a major concern.
NOTE: In bilateral cases, systemic steroid therapy may be preferred to periocular injections. However, in children and adolescents, growth suppression (in addition to the usual complications of long-term systemic steroids) is a major concern.
Transscleral cryotherapy to the area of snowbanking should be considered in patients who fail to respond to either oral or subtenon corticosteroids and who have neovascularization.
Pars plana vitrectomy may be useful in cases refractory to systemic steroids or to treat vitreous opacification, tractional retinal detachment, ERM, and other complications. Additionally, vitreous biopsy through a pars plana vitrectomy may be indicated in cases of suspected masquerade syndromes, especially intraocular lymphoma.
Some physicians delay steroid injections for several weeks to observe whether the IOP increases on topical steroids (steroid response). If a marked steroid response is found, depot injections should be avoided.
Topical NSAIDs are usually not effective in patients with uveitic CME.
Cataracts are a frequent complication of intermediate uveitis. If cataract extraction is performed, the patient should ideally be free of inflammation for 3 months preceding the operation. Consider starting the patient on oral prednisone 60 mg daily 5 days prior to surgery and tapering the prednisone over the next month. Consider a combined pars plana vitrectomy at the time of cataract surgery if significant vitreous opacification is present.
Follow-Up
In the acute phase, patients are reevaluated every 1 to 4 weeks, depending on the severity of the condition.
In the chronic phase, reexamination is performed every 3 to 6 months. Monitor for neovascularization.
12.3 Posterior and Panuveitis
Symptoms
Blurred vision and floaters. Pain, redness, and photophobia are typically absent unless anterior chamber inflammation is present.
Signs
Critical. Cells in the posterior vitreous, vitreous haze, retinal or choroidal inflammatory lesions, retinal vasculitis (sheathing and exudates around vessels).
Other. Anterior and intermediate uveitis (indicative of panuveitis), retinal neovascularization, CME, ERM, and choroidal neovascular membranes.
Differential Diagnosis
Panuveitis
Possible etiologies are listed below:
Sarcoidosis: SEE 12.6, SARCOIDOSIS.
Syphilis: SEE 12.12, SYPHILIS.
VKH syndrome: SEE 12.11, VOGT–KOYANAGI–HARADA SYNDROME.
Lens-induced uveitis: SEE 9.12, LENS-RELATED GLAUCOMA.
Sympathetic ophthalmia: SEE 12.18, SYMPATHETIC OPHTHALMIA.
Tuberculosis: Produces varied clinical manifestations. The diagnosis is usually made by ancillary laboratory tests and response to antituberculosis therapy. Miliary tuberculosis may produce multifocal, small, yellow-white choroidal lesions. Most patients have concomitant anterior granulomatous or nongranulomatous uveitis.
White Dot Syndromes
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE): Acute visual loss in young adults, often after a viral illness. Multiple, creamy yellow–white, plaque-like subretinal lesions in both eyes (see Figures 12.3.1A, B). Lesions block early and stain late on IVFA. Usually spontaneously improves over weeks to months without treatment. May be associated with a cerebral vasculitis (consider MRA if patient has headache or other neurologic symptoms), in which case systemic steroids are indicated.
Multiple evanescent white dot syndrome (MEWDS): Photopsias and acute unilateral visual loss, often after a viral illness and usually in young women. May have a shimmering scotoma. Uncommonly bilateral or sequential. Characterized by multiple, small white lesions deep in the retina or at the level of the retinal pigment epithelium with foveal granularity and occasionally vitreous cells. Fluorescein angiography may show classic perifoveal “wreath-like” pattern. There is often an enlarged blind spot on formal visual field testing. Vision typically returns to normal within weeks without treatment.
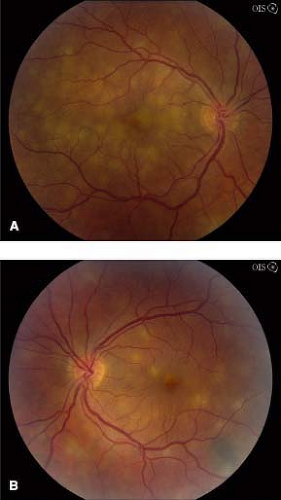
Figure 12.3.1 Fundus photographs of right (A) and left (B) eye showing creamy yellow subretinal lesions in APMPPE. Note a pigmented choroidal nevus along inferotemporal arcade in left eye.
Birdshot retinochoroidopathy: Usually middle-aged patients with bilateral, multiple, creamy-yellow spots deep to the retina, approximately 1 mm in diameter, scattered throughout the fundus but most prominent in inferior quadrants. A mild to moderate vitritis is present. Retinal vasculitis, CME, and optic nerve edema may be present. ICG angiography shows characteristic hypofluorescent spots but fluorescein angiography often shows only retinal vasculitis, CME, and “quenching” of dye. Positive HLA-A29 in 95% to 100% of patients. Early systemic immunosuppression often recommended.
Multifocal choroiditis with panuveitis: Visual loss in young myopic women, typically bilateral. Multiple, small, round, pale inflammatory lesions (similar to histoplasmosis) are located at the level of the pigment epithelium
and choriocapillaris. Unlike histoplasmosis, vitritis occurs in 98% of patients. The lesions can occur in the macula and midperiphery and frequently respond to oral or periocular steroids, but typically recur with tapering, so that immunosuppressive therapy is often necessary. Choroidal neovascularization (CNV) is common, and so patients should return for urgent evaluation if they have decreased vision or metamorphopsia.
Punctate inner choroidopathy: Blurred vision, paracentral scotoma, and/or photopsias, usually in young myopic women. Multiple, small round yellow-white spots predominantly in posterior pole with minimal intraocular inflammation. Lesions become well-demarcated atrophic scars within weeks. CNV may develop in up to 40% of patients. Systemic immunosuppression usually indicated.
Serpiginous choroidopathy: Typically bilateral, recurrent chorioretinitis characterized by acute lesions (yellow-white subretinal patches with indistinct margins) bordering old atrophic scars. The chorioretinal changes usually extend from the optic disc outward; however, one-third may begin in the macula. Patients are typically aged 30 to 60 years. Systemic immunosuppression indicated. CNV may develop. Must be distinguished from “serpiginous” pattern of tuberculous chorioretinitis.
Toxocariasis: Typically unilateral. Usually occurs in children. The most common presentations are a macular granuloma (elevated white retinal/subretinal lesion) with poor vision, unilateral intermediate uveitis with peripheral granuloma, or endophthalmitis. A peripheral lesion may be associated with a fibrous band extending to the optic disc, sometimes resulting in macular vessel dragging. A severe vitritis and anterior uveitis may be present. A negative undiluted Toxocara titer in an immunocompetent host usually rules out this disease. SEE 8.1, LEUKOCORIA.
Presumed ocular histoplasmosis syndrome: Punched-out chorioretinal scars, peripapillary atrophy, and often CNV. Vitreous cells are absent. SEE 11.24, OCULAR HISTOPLASMOSIS.
Retinitis
CMV retinitis: Whitish patches of necrotic retina are mixed with retinal hemorrhage. Vascular sheathing (secondary frosted branch angiitis) in about 20% of eyes. Vitritis and anterior uveitis are usually mild. Seen in immunocompromised patients (most commonly in advanced HIV/AIDS, but also inherited or iatrogenic disorders of the immune system; rarely after periocular or intravitreal steroid injections) and congenitally infected neonates. SEE 12.9, CYTOMEGALOVIRUS RETINITIS.
Acute retinal necrosis (ARN): Unilateral or bilateral peripheral white patches of thickened necrotic retina with vascular sheathing that progress rapidly. Marked vitritis and anterior uveitis are typically present. SEE 12.8, ACUTE RETINAL NECROSIS (ARN).
Progressive outer retinal necrosis (PORN): Clinically similar to ARN, but may not have vitreous cells. Involves the posterior pole or optic nerve early, and classically spares the vessels. Occurs exclusively in severely immunocompromised patients, especially advanced HIV/AIDS, with rapid progression over several days. SEE 12.8, ACUTE RETINAL NECROSIS.
Toxoplasmosis: Unilateral retinal lesion may or may not be associated with an adjacent pigmented chorioretinal scar or clumps of scars. Focal dense vitritis. SEE 12.5, TOXOPLASMOSIS.
Candida: Early discrete drusen-like choroidal lesions progressing to yellow-white, fluffy retinal or preretinal lesions. SEE 12.17, CANDIDA RETINITIS/UVEITIS/ENDOPHTHALMITIS.
Vasculitis
Retinal sheathing around vessels. Branch retinal vein and branch retinal artery occlusions may occur.
Periphlebitis (predominantly veins).
Sarcoidosis: Yellow “candlewax” exudates around veins.
Syphilis.
Pars planitis: Most prominent in the inferior periphery, neovascularization may be present.
Eales disease: Peripheral neovascularization and/or avascular retina.
Multiple sclerosis.
Birdshot retinochoroidopathy.
Arteritis (predominantly arteries).
Giant cell arteritis.
Polyarteritis nodosum.
Frosted branch angiitis.
Churg–Strauss.
ARN.
IRVAN (idiopathic retinal vasculitis, aneurysms, and neuroretinitis).
Susac syndrome.
Both arteries and veins.
Systemic lupus erythematosus.
Granulomatosis with polyangiitis (Wegener granulomatosis).
Behçet disease.
HLA-B27-associated.
Postsurgical/Trauma
Other Infectious Causes of Posterior Uveitis
Cat-scratch disease: Unilateral stellate macular exudates, optic nerve swelling, vitreous cells, positive Bartonella serology. SEE 5.3, PARINAUD OCULOGLANDULAR CONJUNCTIVITIS.
Diffuse unilateral subacute neuroretinitis: Typically unilateral visual loss in children and young adults, caused by a nematode. Optic nerve swelling, vitreous cells, and deep gray-white retinal lesions are present initially, but may be subtle. Later, optic atrophy, narrowing of retinal vessels, and atrophic pigment epithelial changes develop. Vision, visual fields, and ERG deteriorate with time. Treatment is to laser nematode.
Lyme disease: Produces varied forms of posterior uveitis. SEE 13.3, LYME DISEASE.
Nocardia, Coccidioides species, Aspergillus species, Cryptococcus species, meningococcus, ophthalmomyiasis, onchocerciasis, and cysticercosis (seen more commonly in Africa and Central and South America).
Other Causes of Vitreous Cells
Ocular ischemia.
Spillover from anterior uveitis.
Masquerade syndromes: Always consider these in the very old or very young patient.
Large cell lymphoma: Persistent vitreous cells in patients >50 years, which usually do not respond completely to systemic steroids. Yellow-white subretinal infiltrates, retinal edema and hemorrhage, anterior chamber inflammation, or neurologic signs may be present.
Malignant melanoma: A retinal detachment and associated vitritis may obscure the underlying tumor. SEE 11.36, CHOROIDAL NEVUS AND MALIGNANT MELANOMA OF THE CHOROID.
Retinitis pigmentosa: Vitreous cells and macular edema may accompany waxy pallor of the optic disc, “bone-spicule” pigmentary changes, and attenuated retinal vessels. SEE 11.28, RETINITIS PIGMENTOSA AND INHERITED CHORIORETINAL DYSTROPHIES.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


