Purpose
To evaluate the safety, tolerability and bioactivity of ascending doses of MP0112, a designed ankyrin repeat protein (DARPin) that binds with high affinity to vascular endothelial growth factor-A (VEGF-A), in treatment-naive patients with exudative age-related macular degeneration (AMD).
Design
Phase I/II, open-label, multicenter, dose-escalation study.
Methods
Patients were to receive a single intravitreal injection of MP0112 at doses ranging from 0.04 to 3.6 mg and be monitored for 16 weeks for safety, efficacy, pharmacokinetics, and dose response.
Results
Altogether, 32 patients received a single injection of MP0112. The maximum tolerated dose was 1.0 mg because of a case of endophthalmitis in the 2.0 mg cohort. Drug-related adverse events were reported by 13 (41%) of 32 patients; they included ocular inflammation in 11 patients (7 mild, 4 moderate in severity). Visual acuity scores were stable or improved compared with baseline for ≥4 weeks following injection; both retinal thickness and fluorescein angiography leakage decreased in a dose-dependent manner. Rescue therapy was administered to 20 (91%) of 22 patients who received 0.04–0.4 mg MP0112 compared with 4 of 10 (40%) patients who received 1.0 or 2.0 mg. Of patients in the higher-dose cohorts who did not require rescue treatment, 83% (5/6) maintained reductions in central retinal thickness through week 16.
Conclusions
A single injection of 1.0 or 2.0 mg MP0112 resulted in mean decreases in retinal thickness and leakage area despite ocular inflammation. Larger-scale studies are warranted to confirm these observations.
Age-related macular degeneration (AMD) is a leading cause of irreversible central vision loss in people 65 years of age or older. The disease can be subdivided into 2 categories: nonexudative and exudative. Nonexudative AMD is caused by the loss of photoreceptors through the atrophy of retinal pigment epithelium in advanced stages of the disease. Exudative AMD, also termed neovascular AMD, is caused by proliferation of choroidal neovascularization (CNV), leading to bleeding and loss of photoreceptors through fibrovascular scarring. CNV and related manifestations (subretinal hemorrhage, detachment of the retinal pigment epithelium, and fibrovascular disciform scarring) are the most common causes of severe vision loss resulting from AMD. Untreated, exudative AMD can lead to progressive and substantial loss of central vision and a reduction in quality of life.
The relationship between vascular endothelial growth factor-A (VEGF-A) and AMD pathogenesis has led to the development of anti-VEGF therapies that inhibit CNV leakage and reduce vessel permeability. Several VEGF antagonists have been developed, including monoclonal antibodies (ranibizumab and bevacizumab); receptor fragments (aflibercept); and other molecules (pegaptanib, a DNA aptamer). These agents have radically altered the management of neovascular AMD and have become the current standard of care. Anti-VEGF agents are injected directly into the vitreous cavity. Although treatment has evolved from monthly dosing to individualized regimens, the best results are achieved with injections every 4–8 weeks in order to maintain improvement in central vision, placing a considerable burden of treatment on patients, physicians and healthcare systems.
MP0112 is a recombinant protein of the designed ankyrin repeat protein (DARPin) family. DARPins are small, single-domain proteins that can selectively bind to a target protein with high affinity and specificity. These genetically engineered antibody-mimetic proteins show greater stability and at least equal affinity with immunoglobulins, making them effective investigational and therapeutic tools. The in vitro and in vivo effectiveness has been demonstrated in areas that include preclinical tumor targeting and diagnostics.
In vitro, MP0112 has been shown to act as a highly potent antagonist to all VEGF-A isoforms (KD of 1–4 pM; data on file; Molecular Partners, Zurich-Schlieren, Switzerland). Animal studies have demonstrated the high efficacy of MP0112 to inhibit abnormal neovascularization (data on file, Molecular Partners). In a rabbit model of ocular pharmacokinetics with vascular leakage inhibition as read-out, MP0112 was fully active for at least 30 days, whereas ranibizumab did not show activity after 30 days due to faster clearance (data on file, Molecular Partners). Good laboratory-practice toxicology studies were performed and revealed that inflammation can result from potential toxicity in patients (data on file, Molecular Partners).
The mechanism of action of MP0112 is similar to that of ranibizumab, pegaptanib and aflibercept (ie, inhibition of VEGF-A by binding without receptor interaction via the Fc region), and MP0112 has shown a number of advantages compared with the current standard of care: high potency (ie, IC 50 in the low pM range); very long ocular half-life (about 2 weeks) ; and very low systemic exposure (data on file, Molecular Partners). Compared with ranibizumab, MP0112 has greater binding affinity to VEGF-A and is retained in the vitreous for a substantially longer time. The evidence suggests, therefore, that MP0112 has the potential to reduce the frequency of intravitreal injections. A recent study has demonstrated the potential of DARPins compared with currently available agents in DME. The current study was designed to assess the safety, tolerability and preliminary efficacy of intravitreal injections of MP0112 for the treatment of exudative AMD and was performed in parallel with the DME study.
Methods
This phase I/II, open-label, noncontrolled, dose-escalation trial was conducted in 8 ophthalmologic centers in France, Switzerland and the Czech Republic. The study and data accumulation were carried out with approval from the following ethics committees: CPP Ile de France III, Kantonale Ethikkommission Bern, Ethics Committee of Central Military Hospital, Ethics Committee of Faculty Hospital Brno, and Ethics Committee of Faculty Hospital Olomouc. The study adhered to the guidelines of the Declaration of Helsinki, and the protocol and consent forms were approved by a local investigational review board. Each subject provided written consent to participate in this research study. The study is registered at ClinicalTrials.gov under the identifier: NCT01086761.
Study Population
Male and female patients 50 years of age or older who had clinical signs and angiographic evidence of active primary progressive subfoveal CNV, including juxtafoveal lesions that affected the fovea on fluorescein angiography (FA) in the study eye and that were at least 50% of the total lesion area, were eligible for enrollment. Patients were also required to meet the Early Treatment Diabetic Retinopathy Study (ETDRS) best-corrected visual acuity (BCVA) of 70 to 25 letters (Snellen equivalent of 20/40 to 20/320) in the study eye at 4 meters.
Patients with any of the following were excluded from the study: any prior treatment for neovascular AMD in the study eye; a total lesion size of >20 mm 2 ; subretinal hemorrhage either ≥50% of the total lesion area or with ≥2.54 mm 2 blood under the fovea; scar or fibrosis ≥50% of the total lesion in the study eye; or scar, fibrosis or atrophy involving the center of the fovea. Patients with other causes of CNV or ocular surgery (including cataract extraction) in the study eye within 3 months of enrollment were also not eligible to participate.
Study Design
The primary study objective was to assess the safety and tolerability of intravitreal doses of MP0112. Secondary objectives were to assess the preliminary efficacy of MP0112 based on changes in BCVA, central retinal thickness (CRT) as measured by optical coherence tomography (OCT), and CNV leakage as measured by FA. Additionally, the systemic pharmacokinetic profile and immunogenicity of intravitreally administered MP0112 were assessed.
The study was designed in 2 stages. Part A consisted of a dose-escalation design in which 6 cohorts received a single MP0112 dose of 0.04 mg, 0.15 mg, 0.4 mg, 1.0 mg, 2.0 mg, or 3.6 mg. Patients were enrolled into the study sequentially. The first patient in each dose cohort received a single intravitreal injection of MP0112 in 1 eye. If no severe or serious ocular adverse event (AE) that was considered to be drug related occurred within 2 weeks of administration, the remaining 5 patients in the dose cohort were recruited and dosed. Dose escalation proceeded only (1) after all patients in a dose cohort had received the specified dose; (2) if moderate ocular toxicity, as defined by the protocol, affected no more than 2 of 6 patients within the dosing cohort after a minimum follow-up of 1 week; and (3) if the Medical Review Committee had approved the dose escalation.
Study Treatments and Procedures
MP0112 was administered as a single intravitreal injection (0.05 mL) using a 30-gauge needle and standard techniques, including the use of a lid speculum, topical anesthesia and 5% povidone-iodine. All patients remained under observation in the clinic for up to 5 hours after dosing. Patients were examined before and after injection and received a safety follow-up call the day after dosing, with referral to an ophthalmologist if required. Follow-up visits were made 3 days, and 1, 2, 4, 8, 12, and 16 weeks after treatment. At day 3, patients underwent a complete eye exam (including slit-lamp biomicroscopy and indirect ophthalmoscopy) and pharmacokinetic assessment. At each study visit, patients were assessed for AEs, concomitant medications, pharmacokinetics (until week 12), complete eye exams, BCVA and OCT. FA was assessed at baseline and week 4 ( Figure 1 ).
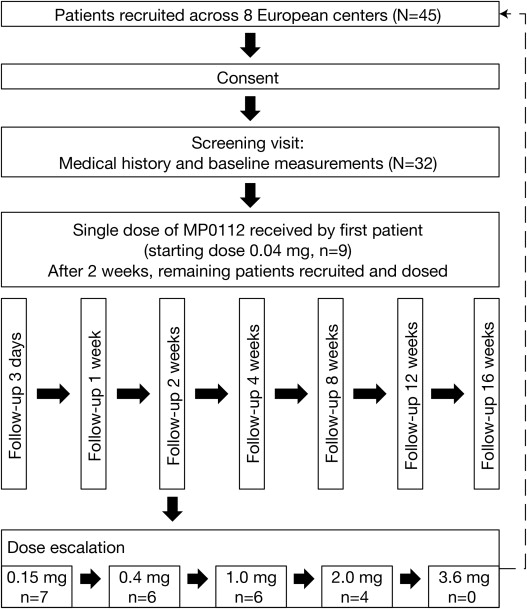
At the investigators’ discretion, patients could be given rescue therapy with standard-of-care treatments from 2 weeks after administration of MP0112. The criteria for initiation of rescue therapy differed slightly by region: in the Czech Republic and France, patients were eligible for rescue therapy if they experienced at least 1 of the following: visual acuity (VA) deterioration of ≥6 letters from baseline; an increase in lesion size or leakage; the formation of new lesions; or an increase in subretinal fluid. In Switzerland, rescue therapy applied to patients who experienced VA deterioration of ≥6 letters from baseline or a decrease in CRT of <50 μm from baseline. All patients, including those who received rescue therapy, were followed for 16 weeks.
Optical coherence tomography
OCT was performed at each study site using Stratus OCT 3 (Carl Zeiss Meditec, Jena, Germany) and Spectralis OCT (Heidelberg Engineering, Heidelberg, Germany), if available. The same OCT unit was used for all visits for a given patient so as to allow for comparison among visits. Stratus OCT data were used for comparison across all sites because Spectralis OCT data were not available for all patients. The central subfield thickness, which is the average thickness within the central 1 mm of the fovea, was used as a measure of CRT for all OCT devices. Scans were acquired using the fast macular scan protocol on Stratus (Carl Zeiss Meditec), which consists of 6-line B-scans (each consisting of 128 A-scans per line), each 6 mm long, centered on the fixation point and spaced 30 degrees apart around a circle. Scans were acquired using the high-speed spectral-domain OCT volume mode on the Heidelberg Spectralis, which consists of 25 horizontal-line B-scans (each consisting of 512 A-scans per line; the line scans were saved for analysis after 9 frames and averaged) covering a total area of 20 × 20 degrees of the macula with a distance of 240 μm between the horizontal lines. OCT images were analyzed and graded by the Central Reading Center (Bern Photographic Reading Center, Bern, Switzerland).
Fluorescein angiography
Digital images at the 30- to 40-degree setting (depending on the device) were taken using the Heidelberg HRA System (Heidelberg Engineering); MRP OphthaVision (MRP Group, Waltham, Massachusetts, USA); Ophthalmic Imaging Systems (OIS) WinStation (Sacramento, California, USA); Topcon IMAGEnet (Capelle a/d Ijssel, Netherlands); or Zeiss Visupac digital systems (Carl Zeiss Meditec). The fluorescein angiogram contained stereoscopic views of 2 fields at specified times (up to 10 minutes) after fluorescein injection. These fields included the macula (ETDRS Field 2) of both eyes and the disc field (ETDRS Field 1M) of the study eye. Stereoscopic red-free photographs were taken of ETDRS Field 2 in each eye prior to the injection of the fluorescein dye. FA images were analyzed and graded by the Central Reading Center (Bern Photographic Reading Center).
Statistical analysis
No formal significance or analytic testing was performed due to the small sample size. Continuous variables were summarized using descriptive statistics, and categoric variables were described using counts and percentages.
Results
Baseline Patient Characteristics
Of the 45 patients screened, 32 met the inclusion/exclusion criteria and received a single intravitreal injection of MP0112 in the study eye (0.04 mg, 9 patients; 0.15 mg, 7 patients; 0.4 mg, 6 patients; 1.0 mg, 6 patients; 2.0 mg, 4 patients). All 32 patients completed the study. The baseline characteristics of the study population are summarized in Table 1 .
| MP0112 (n = 32) | |
|---|---|
| Age (years) | |
| Mean ± SD | 78.3 ± 5.3 |
| Range | 65, 87 |
| Gender, n (%) | |
| Male | 11 (34%) |
| Female | 21 (66%) |
| Ethnicity, n (%) | |
| Caucasian/white | 31 (97%) |
| Ocular measures | |
| Classic CNV, n (%) a | 8 (25%) |
| Occult CNV, n (%) a | 29 (91%) |
| VA score, mean ± SD (range) | 58.7 ± 11.6 letters (32–72) |
| CRT, mean ± SD (range) | 351.5 ± 107.8 μm (191–790) |
a One patient could not be graded, and another patient could not be graded for classic CNV.
Safety
AEs that were considered to be drug related were reported in13 of 32 (41%) patients and included anterior chamber inflammation (5/13 patients); vitritis (4/13 patients); anterior chamber cell flare (3/13 patients); and endophthalmitis (1/13) ( Table 2 ). Ocular inflammation resolved without consequence in all eyes; in 36% (4/11), this occurred without treatment, and all others received local anti-inflammatory medication (betamethasone, dexamethasone, tropicamide, or dexamethasone-tobramycin).
| MP0112 Dose Cohort | |||||
|---|---|---|---|---|---|
| 0.04 mg n = 9 | 0.15 g n = 7 | 0.4 mg n = 6 | 1.0 mg n = 6 | 2.0 mg n = 4 | |
| Vitritis | 0 | 1 (14%) | 0 | 2 (33%) | 1 (25%) |
| Anterior chamber inflammation | 0 | 1 (14%) | 1 (17%) | 2 (33%) | 1 (25%) |
| Anterior chamber cell flare | 0 | 0 | 1 (17%) | 0 | 2 (50%) |
∗ Patients received a single intraocular injection of MP0112.
One serious AE (3%) was reported during the study: a patient who received 2.0 mg MP0112 developed severe ocular inflammation with hypopyon, initially classified as endophthalmitis. The patient received a 2-day course of intravenous vancomycin and ceftriaxone, oral prednisolone, and Kefzol eye drops. The hypopyon was completely resolved within 3 days from onset. No Gram staining or cultures were performed, but the mild course and response to steroids suggest that sterile endophthalmitis had occurred. Based on this severe ocular inflammation, the maximum tolerated dose was determined to be 1.0 mg. A second stage of the study that was planned to evaluate repeat doses of MP0112 was not initiated because ocular inflammation was observed and was attributed to impurities in the investigative product.
AEs noted by the investigator to be related to the procedure were reported in 3 of 32 (9%) patients (conjunctival hemorrhage, vitreous detachment and hypertension, each occurring in 1 patient).
Antidrug antibodies were detected in the serum of 8 patients. No further characterization of these was performed.
Efficacy
Optical coherence tomography
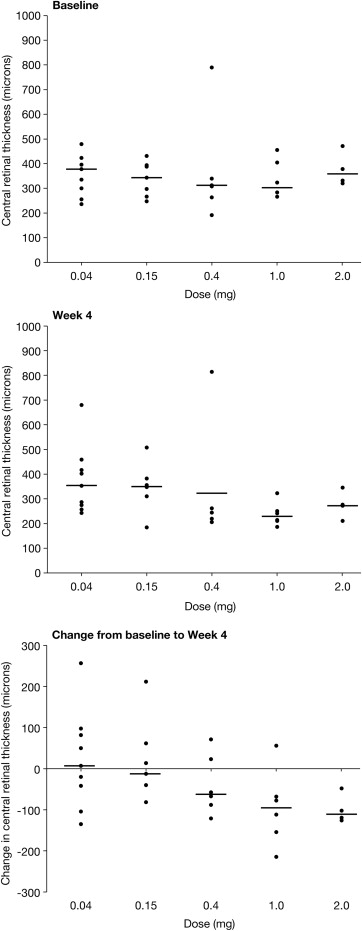
The mean and median CRTs at baseline were 352 μm and 334 μm, respectively (standard deviation, 107.8 μm; range, 191–790) ( Table 1 ). Generally, the higher-dose cohorts experienced a greater decrease in CRT during the 4-week study period ( Figure 2 ). Patients who received 1.0 and 2.0 mg of MP0112 showed the greatest median reductions at week 4 of −95 μm and −111 μm, respectively, compared with 7 μm, −12 μm, and −62 μm in patients who received 0.04 mg, 0.15 mg, and 0.4 mg, respectively. The overall change in CRT across the dosing cohorts is shown in Figure 2 .
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


