Purpose
To estimate the incidence of trilateral retinoblastoma in patients with retinoblastoma.
Design
Systematic review and meta-analysis.
Methods
We searched Medline and Embase for scientific literature published between January 1966 and July 2015 that assessed trilateral retinoblastoma incidence. We used a random-effects model for the statistical analyses.
Results
We included 23 retinoblastoma cohorts from 26 studies. For patients with bilateral retinoblastoma the unadjusted chance of developing trilateral retinoblastoma across all cohorts was 5.3% (95% confidence interval [CI]: 3.3%–7.7%); the chance of pineal trilateral retinoblastoma was 4.2% (95% CI: 2.6%–6.2%) and the chance of nonpineal trilateral retinoblastoma was 0.8% (95% CI: 0.4%–1.3%). In patients with hereditary retinoblastoma (all bilateral cases, and the unilateral cases with a family history or germline RB1 mutation) we found a trilateral retinoblastoma incidence of 4.1% (95% CI: 1.9%–7.1%) and a pineal trilateral retinoblastoma incidence of 3.7% (95% CI: 1.8%–6.2%). To reduce the risk of overestimation bias we restricted analysis to retinoblastoma cohorts with a minimum size of 100 patients, resulting in adjusted incidences of 3.8% (95% CI: 2.4%–5.4%), 2.9% (95% CI: 1.9%–4.2%), and 0.7% (95% CI: 0.3%–1.2%) for any, pineal, and nonpineal trilateral retinoblastoma, respectively, among patients with bilateral retinoblastoma. Among hereditary retinoblastoma we found an adjusted trilateral retinoblastoma incidence of 3.5% (95% CI: 1.2%–6.7%) and a pineal trilateral retinoblastoma incidence of 3.2% (95% CI: 1.4%–5.6%).
Conclusion
The estimated incidence of trilateral retinoblastoma is lower than what is reported in previous literature, especially after exclusion of small cohorts that were subject to overestimation bias in this context.
Until the age of about 7 years patients with hereditary retinoblastoma are at risk of having an intracranial midline primitive neuroectodermal tumor diagnosed, and among patients diagnosed since 1995 more than 95% have developed trilateral retinoblastoma before the age of 5 years. In histopathologic analysis these tumors look similar to corresponding retinal tumors. When unilateral or bilateral retinoblastoma and an intracranial midline primitive neuroectodermal tumor both occur in a patient, this is referred to as trilateral retinoblastoma, which can be found in the pineal gland (pineal trilateral retinoblastoma) in about three-quarters of cases; in the remaining patients it develops in other midline brain regions (nonpineal trilateral retinoblastoma), usually the suprasellar and parasellar region, although other brain regions have been reported also.
In a recent meta-analysis we showed that survival has improved considerably in the last 2 decades, from hardly any to almost half of all patients. Favorable survival after pineal trilateral retinoblastoma depended strongly on early detection and small tumor size. The improved survival after trilateral retinoblastoma was highly associated with the use of (improved) chemotherapy regimens, especially high-dose chemotherapy with stem cell rescue.
Previous radiotherapy for retinoblastoma, especially before the age of 12 months, has been associated with a potentially higher incidence of pineal trilateral retinoblastoma in patients with hereditary retinoblastoma, even though the pineal gland is usually not (directly) within the field of radiation. Whether previous systemic chemotherapy is protective of developing trilateral retinoblastoma is still being debated.
There have been numerous reports on trilateral retinoblastoma incidence, but these studies are quite heterogeneous. Some are referral-based, others population-based. The only previously published study summarizing incidence data across studies reported an incidence of 5%–15% among patients with bilateral retinoblastoma.
The objective of this study is to provide an overview of, to critically analyze, and to provide pooled summary estimates of published incidence data for pineal and nonpineal trilateral retinoblastoma.
Methods
We performed this systematic review and meta-analysis according to the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) statement.
Search Strategy
We searched Medline (PubMed) and Embase for English, Dutch, and German literature published from January 1, 1966 through July 15, 2015, evaluating trilateral retinoblastoma cases. We also considered alternatively found studies for inclusion (eg, through checking references in included studies). The search was similar to the search we used in our systematic review and meta-analysis on survival of patients with trilateral retinoblastoma. To ensure a sensitive search, we included only keywords corresponding to the target condition (including retinoblastoma, pineoblastoma, pineal, suprasellar, parasellar, sellar, ectopic, and brain), without any delimiters. For the detailed search see Supplemental Table 1 (available at AJO.com ).
Study Selection and Data Extraction
Two authors (M.C.J. and A.C.M.) independently reviewed article titles and abstracts for eligibility. Discrepancies were resolved by consensus. Subsequently the same 2 authors independently reviewed eligible full-text articles for inclusion in the systematic review and meta-analysis. Again, discrepancies were solved by consensus.
We included studies in the systematic review and meta-analysis if (1) the study mentioned the number of patients with trilateral retinoblastoma (can also be 0, as long as authors mentioned evaluating for trilateral retinoblastoma cases), if (2) articles reported details and size of the retinoblastoma cohort in which patients with trilateral retinoblastoma were seen (eg, number of patients with trilateral retinoblastoma diagnosed in a cohort of patients with retinoblastoma during a certain period), and if (3) the full-text article could be obtained. We excluded studies from the systematic review if (1) the article was a review or meta-analysis, and if (2) studies included overlapping incidence data. Two authors (M.C.J. and W.A.K.) independently extracted incidence data. Discrepancies were resolved by consensus. We have defined hereditary retinoblastoma as patients with bilateral retinoblastoma, known familial retinoblastoma, or a detected germline mutation in RB1 .
Data Synthesis and Statistical Analysis
Ideally, we would present the cumulative incidence of trilateral retinoblastoma until a certain age (eg, up to 5 years) in comparable retinoblastoma cohorts; however, most studies only provided the total number of patients with retinoblastoma and the number of trilateral retinoblastoma cases they found in their cohort. Therefore we are restricted to the calculation of a percentage of cases that developed trilateral retinoblastoma divided by total retinoblastoma cohort from a certain time period. This method unfortunately does not take into account the effect of patients lost to follow-up (ie, assuming that of all patients with retinoblastoma it is unequivocally known whether they developed trilateral retinoblastoma or not).
In addition to performing unadjusted analysis, we calculated adjusted estimates by including cohorts of at least 100 patients with retinoblastoma, to prevent overestimation bias. Most small cohorts published specifically after encountering at least 1 trilateral retinoblastoma and thus led to a report irrespective of how many patients without trilateral retinoblastoma had been seen (ie, an arbitrary cohort of patients with retinoblastoma with a certain start and end date will have been constructed around the patient(s) with trilateral retinoblastoma that were encountered). The limit of 100 patients was based on visual evaluation of a plot of the incidence of trilateral retinoblastoma against the total sample size across studies ( Figure 1 ). Typically, an unselected patient population contains 40% of bilateral retinoblastoma, and thus we considered also a cohort of at least 40 bilateral retinoblastomas to be large enough to guard for overestimation bias.
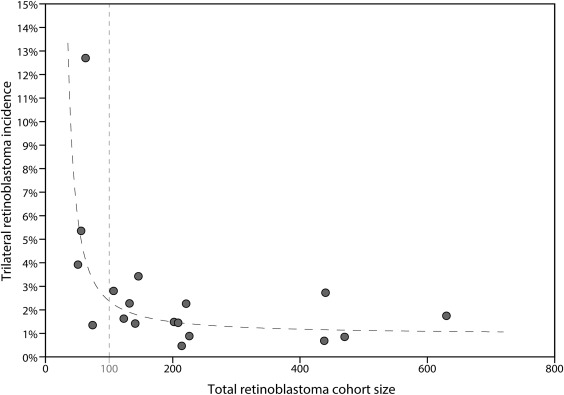
To assess the extent of this bias we compared data from developed vs developing countries, as one would expect follow-up to be more complete in developed countries (ie, higher chance to find a trilateral retinoblastoma). This might lead to an underestimation of trilateral retinoblastoma incidence in the developing countries. Pooling data from different retinoblastoma cohorts also assumes comparability of these cohorts.
To evaluate changes in trilateral retinoblastoma incidence over time we used a cutoff at the year 1995. We chose this cutoff because around (or maybe even before) 1995 treatment of retinoblastoma started to shift from radiotherapy to chemotherapy.
We used a random-effects model to calculate summary estimates of trilateral retinoblastoma incidence. A random-effects model is used for meta-analyses to account for heterogeneity between studies. For each analysis we calculated I 2 to evaluate heterogeneity among included studies; I 2 ranges from 0 to 100% with increasing heterogeneity. For the random-effects analyses we used MetaXL (version 2.1; EpiGear, Wilston, Australia) and SAS (Proc MIXED, version 9.3; SAS, Cary, North Carolina, USA). The forest plots were created with Illustrator CS6 (Adobe, San Jose, California, USA).
Risk of Bias and Study Quality Assessment
Two authors (M.C.J. and T.K.) assessed the risk of bias with a modified checklist that was developed for prevalence studies by Hoy and associates. With the checklist 6 items that could lead to bias were assessed ( Supplemental Table 2 , available at AJO.com ).
Results
We identified 1865 unique studies from database searches. We excluded 1734 articles based on title and abstract ( Figure 2 ). We excluded 105 studies based on the full text; reasons for exclusion are shown in Figure 2 . Twenty-six studies (3 pairs of studies had overlapping cohorts but provided different data of interest and were therefore included; see Table 1 ) met the inclusion criteria of this systematic review. Twenty-one studies (20 cohorts) were included in the meta-analysis. Tables 2 and 3 show the incidence data as reported in each included study.
| Study | Inclusion Period | Country | Source of the Data | Type of Patients With Retinoblastoma | Age at Retinoblastoma Diagnosis (Months) | Patient Follow-up From Retinoblastoma Diagnosis (Months) | Percentage of Cohort With Unilateral Retinoblastoma | Percentage of Cohort With Bilateral Retinoblastoma | Percentage of Cohort With Familial Retinoblastoma | Study Cohort (Partly) Overlaps With |
|---|---|---|---|---|---|---|---|---|---|---|
| Amoaku et al | 1960–1994 | United Kingdom | Population registry | Any | Mean 6, range 0.75–17 | 64% | 36% | 38 a | ||
| Antoneli et al | 1986–2003 | Brasil | 1 center | Any | 60% | 40% | ||||
| Azar et al | 1975–2001 | Australia | 2 centers | Any | Mean 17.9 (bilateral 22.6, unilateral 3.5) | 60% | 40% | 11 | ||
| Bartuma et al | 2001–2011 | Sweden | 1 center | Hereditary | Mean 8, range 0–39 | Mean 60, range 13–144 | 8% | 92% | 38 | |
| Blach et al | 1979–1990 | USA | 1 center | Irradiated | Median 7, range <1–60 | Median 68, range 4–153 | 17% | 83% | 27 | |
| Chantada et al | 1988–2012 | Argentina | 1 center | Bilateral | Mean 13.9, range 0–114 | Median 115, range 31–290 | 100% | 14 a | Moreno et al | |
| De Ioris et al | 1999–2009 | Italy | 1 center | Any | Median 10, range 0.5–73 | 58% | 42% | 14 | ||
| De Potter et al | 1972–1992 | United Kingdom | 1 center | Any | 54% | 46% | 14 | |||
| Duncan et al | 1990–1998 | USA | 2 centers | Any | Mean 44.8, range 0–139 | ≥63% | ≤37% | |||
| Helveston et al | 1967–1987 | USA | 1 center | Any | Unilateral 23, bilateral 10 b | 59% | 41% | |||
| Imhof et al | 1971–1993 | Netherlands | 1 center | Irradiated | Mean 5, range 1–216 | Mean 148, range 48–276 | Moll et al | |||
| Jubran et al | 1991–1999 | USA | 1 center | Any | ||||||
| Jurkiewicz et al | 1996–2008 | Poland | 1 center | Any | Unilateral median 22, bilateral median 12 | |||||
| Kingston et al | 1954–1983 | United Kingdom | 2 centers | Any | 31% | 69% | ||||
| Klufas et al | 2006–2010 | USA | 1 center | Treated with intraarterial chemotherapy | Median 17.5, range 8–36 | |||||
| Lim et al | 2001–2009 | Malaysia | 1 center | Any | ||||||
| Lim et al | 1997–2010 | Singapore | 1 center | Any | 25.7, SD 19.9 (unilateral mean: 30.2, SD 21.4, bilateral mean: 15.4, SD 9.6) | Median 36, range 0–156 | 69% | 31% | ||
| Lueder et al | 1924–1985 | USA | 1 center | Any | 67% | 33% | 14 | Lueder et al | ||
| Lueder et al. | 1924–1989 | USA | 1 center | Any | 14 | Lueder et al | ||||
| Moll et al | 1970–1997 | Netherlands | Population registry | Hereditary | Imhof et al | |||||
| Moreno et al | 2000–2009 | Argentina | Population registry | Any | Unilateral median 26 (IQR 13–42), bilateral median 10 (IQR 5–19) | 68% | 32% | Chantada et al | ||
| Popovic et al | 1990–2001 | Switzerland | 1 center | Any | 49% | 51% | ||||
| Provenzale et al | 1985–2002 | USA | 1 center | Any | 52% | 48% | ||||
| Ramasubramanian et al | 2000–2012 | USA | 1 center | Any | Mean 21, median 13, range 0–91 | 53% | 47% | 11 | ||
| Scott et al | 1970–1990 | USA | 1 center | Any | Mean 15.8 (nonhereditary mean 20.5, hereditary mean 11.7) | 52% | 48% | |||
| Shields et al | 1995–1999 | USA | 1 center | Any | Mean 14, median 8, range 1–87 | Mean 33, range 0–67 | 48% | 52% | 18 |
a As percentage of the patients with bilateral retinoblastoma.
b Age at treatment; unknown if these are means or medians or some other measure.
| Study | Inclusion Period | Unilateral and Bilateral Retinoblastoma | Bilateral Retinoblastoma | Hereditary Retinoblastoma | |||
|---|---|---|---|---|---|---|---|
| Pineal | Nonpineal | Pineal | Nonpineal | Pineal | Nonpineal | ||
| Amoaku et al | 1960–1994 | 2.7% (4/146) | 0.7% (1/146) | 7.7% (4/52) | 1.9% (1/52) | ||
| Antoneli et al | 1986–2003 | 0.6% (3/470) | 0.2% (1/470) | 1.1% (2/186) | 0.5% (1/186) | ||
| Azar et al | 1975–2001 | 1.6% (2/123) | 0.0% (0/123) | 4.1% (2/49) | 0.0% (0/49) | ||
| De Ioris et al | 1999–2012 | 2.8% (3/107) | 0.0% (0/107) | 6.7% (3/49) | 0.0% (0/49) | ||
| De Potter et al | 1972–1992 | 2.0% (9/440) | 0.7% (3/440) | 4.0% (8/202) | 1.5% (3/202) | ||
| Duncan et al | 1990–1998 | 0.9% (2/226) | 0.0% (0/226) | 2.4% (2/85) a | 0.0% (0/85) a | ||
| Helveston et al | 1967–1987 | 1.4% (1/74) | 0.0% (0/74) | 3.3% (1/30) | 0.0% (0/30) | ||
| Jubran et al | 1991–1999 | 1.4% (3/207) | 0.0% (0/207) | ||||
| Jurkiewicz et al | 1996–2008 | 0.5% (1/202) | 1.0% (2/202) | ||||
| Kingston et al | 1954–1983 | 1.3% (8/630) | 0.5% (3/630) | 1.9% (8/432) | 0.5% (2/432) | ||
| Lim et al | 2001–2009 | 1.4% (2/141) | 0.0% (0/141) | ||||
| Lim et al | 1997–2010 | 3.9% (2/51) | 0.0% (0/51) | 12.5% (2/16) | 0.0% (0/16) | ||
| Lueder et al | 1924–1985 | 2.3% (3/132) | 0.0% (0/132) | 6.8% (3/44) | 0.0% (0/44) | 6.0% (3/50) | 0.0% (0/50) |
| Lueder et al | 1924–1989 | 2.7% (4/143) | 7.1% (4/56) | ||||
| Moll et al | 1970–1997 | 5.8% (7/121) | 0.0% (0/121) | ||||
| Moreno et al | 2000–2009 | 0.7% (3/438) | 0.0% (0/438) | 2.2% (3/139) | 0.0% (0/139) | ||
| Popovic et al | 1990–2001 | 1.8% (4/221) | 0.5% (1/221) | 3.7% (4/108) | 0.9% (1/108) | ||
| Provenzale et al | 1985–2002 | 11.1% (7/63) | 1.6% (1/63) | 23.3% (7/30) | 3.3% (1/30) | ||
| Ramasubramanian et al | 2000–2012 | 1.0% (4/408) | 1.6% (3/193) | 1.9% (4/215) | |||
| Scott et al | 1970–1990 | 5.4% (3/56) | 0.0% (0/56) | 11.1% (3/27) | 0.0% (0/27) | 10.0% (3/30) | 0.0% (0/30) |
| Shields et al | 1995–1999 | 0.5% (1/214) | 0.0% (0/214) | 0.9% (1/112) | 0.0% (0/112) | 0.9% (1/117) | 0.0% (0/117) |
a Duncan et al reported that 83 had hereditary retinoblastoma, excluding the 2 pineal trilateral retinoblastoma cases that can be classified as having hereditary retinoblastoma on the basis of developing a midline primitive neuroectodermal tumor (they also presented a case with an “orbital midline primitive neuroectodermal tumor,” but we are not convinced this is trilateral retinoblastoma).
| Study | Inclusion Period | Treatment for Retinoblastoma | Unilateral and Bilateral Retinoblastoma | Bilateral Retinoblastoma | Hereditary Retinoblastoma | |||
|---|---|---|---|---|---|---|---|---|
| Pineal | Nonpineal | Pineal | Nonpineal | Pineal | Nonpineal | |||
| Bartuma et al | 2001–2011 | Systemic chemotherapy | 0.0% (0/24) a | 0.0% (0/24) a | ||||
| Chantada et al | 1988–2009 | Systemic chemotherapy | 1.9% (3/159) | 0.0% (0/159) | ||||
| No systemic chemotherapy | 0.0% (0/38) | 0.0% (0/38) | ||||||
| Klufas et al | 2006–2010 | Intraarterial chemotherapy | 1.1% (1/89) | 0.0% (0/89) | 1.4% (1/70) | 0.0% (0/70) | ||
| Ramasubramania et al | 2000–2012 | Systemic chemotherapy | 0.4% (1/252) | 0.6% (1/180) | ||||
| No systemic chemotherapy | 1.9% (3/156) | 8.6% (3/35) | ||||||
| Shields et al | 1995–1999 | Systemic chemotherapy | 0.0% (0/142) | 0.0% (0/142) | 0.0% (0/95) | 0.0% (0/95) | 0.0% (0/99) | 0.0% (0/99) |
| No systemic chemotherapy | 1.4% (1/72) | 0.0% (0/72) | 5.9% (1/17) | 0.0% (0/17) | 5.6% (1/18) | 0.0% (0/18) | ||
| Blach et al | 1979–1990 | Radiotherapy | 4.3% (5/117) | 0.9% (1/117) | 5.2% (5/97) | 1.0% (1/97) | ||
| Imhof et al | 1971–1993 | Radiotherapy | 4.7% (5/106) | 0.0% (0/106) | 5.7% (5/87) | 0.0% (0/87) | ||
| Ramasubramanian et al | 2000–2012 | Radiotherapy | 2.8% (1/87) | |||||
| No radiotherapy | 1.7% (3/179) | |||||||
a Twenty-four patients received a full course of systemic chemotherapy, 1 was previously treated elsewhere, and 2 did not receive (a full course of) chemotherapy; 1 of these latter 2 patients did develop trilateral retinoblastoma (location unspecified).
Unadjusted Estimates
Twenty-six studies reported trilateral retinoblastoma incidence in 23 unique cohorts of patients with retinoblastoma, and in 15 studies (15 cohorts) bilateral could be distinguished from unilateral retinoblastoma ( Table 2 ). Seven studies (6 cohorts) presented the trilateral retinoblastoma incidence in hereditary retinoblastoma, 3 studies (cohorts) reported trilateral retinoblastoma incidence after external beam irradiation for retinoblastoma, and 5 studies (cohorts) compared the incidence in patients with retinoblastoma with and without previous chemotherapy. Forest plots of the included studies (sorted by the midpoint of the study period) and the summary estimates are shown in Figure 3 .
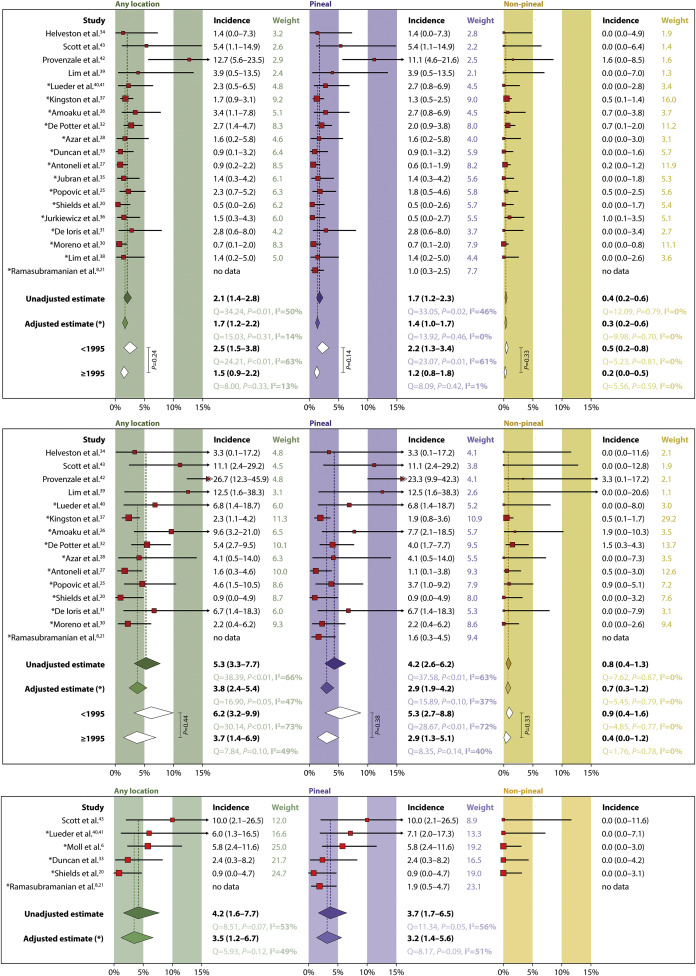
For unilateral and bilateral retinoblastoma combined, the unadjusted chance of developing a trilateral retinoblastoma across all studies is 2.1% (95% confidence interval [CI]: 1.4%–2.8%; 18 cohorts), the chance of developing pineal trilateral retinoblastoma is 1.7% (95% CI: 1.2%–2.3%; 19 cohorts), and the chance of a nonpineal trilateral retinoblastoma is 0.4% (95% CI: 0.2%–0.6%; 18 cohorts). For bilateral retinoblastoma the chance of developing a trilateral retinoblastoma is 5.3% (95% CI: 3.3%–7.7%; 14 cohorts); restricting calculations to pineal trilateral retinoblastoma resulted in an incidence of 4.2% (95% CI: 2.6%–6.2%; 15 cohorts) and restricting to nonpineal trilateral retinoblastoma gave an incidence of 0.8% (95% CI: 0.4%–1.3%; 14 cohorts). In hereditary retinoblastoma we found a trilateral retinoblastoma incidence of 4.2% (95% CI: 1.6%–7.7%; 5 cohorts) and a pineal trilateral retinoblastoma incidence of 3.7% (95% CI: 1.8%–6.2%; 6 cohorts), and we did not calculate the nonpineal trilateral retinoblastoma incidence, as there were no cases in 5 retinoblastoma cohorts.
Adjusted Estimates
We adjusted for potential overestimation bias by restricting the analysis to cohorts that included at least 100 patients with retinoblastoma ( Figure 1 ). We found incidences of 1.7% (95% CI: 1.2%–2.2%; 14 cohorts), 1.4% (95% CI: 1.0%–1.7%; 15 cohorts), and 0.3% (95% CI: 0.2%–0.6%; 14 cohorts) for any, pineal, and nonpineal trilateral retinoblastoma, respectively. In cohorts with only patient with bilateral retinoblastoma we found incidences of 3.8% (95% CI: 2.4%–5.4%; 10 cohorts), 2.9% (95% CI: 1.9%–4.2%; 11 cohorts) and 0.7% (95% CI: 0.3%–1.2%; 10 cohorts), respectively. Among patients with hereditary retinoblastoma we found a trilateral retinoblastoma incidence of 3.5% (95% CI: 1.2%–6.7%; 4 cohorts) and a pineal trilateral retinoblastoma incidence of 3.2% (95% CI: 1.4%–5.6%; 5 cohorts).
Period Analysis
To analyze changes over time we created 2 groups with the year 1995 as the cutoff year (depending on the midpoint of the study period). Before the year 1995 unadjusted trilateral retinoblastoma incidence for unilateral and bilateral retinoblastoma combined was 2.5% (95% CI: 1.5%–3.8%) vs 1.5% (95% CI: 0.9%–2.2%) from the year 1995 onward ( P = .24). The incidence of pineal trilateral retinoblastoma before the year 1995 was 2.2% (95% CI: 1.3%–3.4%) vs 1.2% (95% CI: 0.8%–1.8%) from 1995 onward ( P = .14).
Restricted to patients with bilateral retinoblastoma, the unadjusted trilateral retinoblastoma incidence was 6.2% (95% CI: 3.2%–9.9%) before the year 1995 vs 3.7% (95% CI: 1.4%–6.9%) from the year 1995 onward ( P = .44). The incidence of pineal trilateral retinoblastoma was 5.3% (95% CI: 2.7%–8.8%) before the year 1995 vs 2.9% (95% CI: 1.3%–5.1%) from the year 1995 onward ( P = .38; Figure 3 ).
Effect of Previous Therapy
Four studies reported on trilateral retinoblastoma incidence in retinoblastoma cohorts who underwent previous systemic chemotherapy ( Table 3 ). In a single-center study with 4 trilateral retinoblastoma cases an inverse association between chemotherapy and the development of pineoblastoma was reported ( P = .014), with an incidence of 0.6% (1/180) and 8.6% (3/35), respectively, for patients who did and who did not receive previous chemotherapy for their retinoblastoma. In a single-center study from the same center, but different period, 1 pineoblastoma was found in 18 hereditary patients with retinoblastoma who did not undergo chemotherapy, compared to none in 99 patients who did undergo chemotherapy. However, in another single-center study with 3 trilateral retinoblastomas this association was reversed with an incidence of 1.9% (3/159) vs 0.0% (0/38), respectively ( P > .99).
Three studies specifically looked at trilateral retinoblastoma incidence in cohorts of patients with retinoblastoma who underwent external beam radiotherapy ( Table 3 ). In a single-center study with 4 trilateral retinoblastomas a pineal trilateral retinoblastoma incidence of 1.7% (3/179) was found in the group of patients with nonirradiated hereditary retinoblastoma and an incidence of 2.8% (1/36) was found in the irradiated group ( P = .5). A single-center study with 6 trilateral retinoblastomas found that patients with bilateral retinoblastoma who underwent irradiation as treatment for their retinoblastoma had a 6.2% (6/97) chance to develop trilateral retinoblastoma (5 pineal and 1 suprasellar); excluding the suprasellar tumor from the calculation resulted in a pineal trilateral retinoblastoma incidence of 5.2% (5/97). Finally, in a single-center study with 5 cases a pineal trilateral retinoblastoma incidence of 5.7% (5/87) in patients with irradiated hereditary retinoblastoma was reported.
Risk of Bias and Study Quality Assessment
Per-study scores on individual items of the risk-of-bias checklist (numbered from Q1 through Q6) can be found in Supplemental Table 2 (available at AJO.com ), showing considerable risk of bias in terms of how much the cohort is population-like (Q1 and Q2) and the follow-up duration (Q4). To address assessment bias we compared trilateral retinoblastoma incidence in developed vs developing countries—the latter being potentially more prone to this type of bias, which would result in lower expected incidence numbers. This comparison indeed showed differences with unadjusted unilateral and bilateral trilateral retinoblastoma incidences of 2.3% (95% CI 1.6%–3.2%) vs 1.1% (95% CI 0.5%–1.9%; P = .32) and bilateral trilateral retinoblastoma incidences of 6.0% (95% CI 3.5%–9.2%) vs 2.6% (95% CI 0.4%–6.2%; P = .50) for developed and developing countries, respectively, though statistically not significantly different.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


