Stromal and Pre-Descemet’s Dystrophies
Puwat Charukamnoetkanok
Shu-Wen Chang
Dimitri T. Azar
Stromal and pre-Descemet’s dystrophies are characterized by the appearance of deposits within or between the stromal keratocytes. The most common stromal dystrophies (granular, lattice, and macular dystrophies) often result in progressive visual loss (1). In contrast, pre-Descemet’s dystrophy and other less common stromal dystrophies (such as fleck dystrophy and central cloudy dystrophy) seldom result in decreased vision. The classification of stromal dystrophies was traditionally based on the clinical appearance of the stromal deposits and on the immunohistological and ultrastructural findings. Recent advances in molecular genetics have better clarified the similarities among various dystrophies (2). For example, granular, lattice, Avellino, and Reis-Bücklers dystrophies result from defects in the same gene on chromosome 5q31 (keratoepithelin gene). New technologic advances also lead to new treatment options for anterior stromal dystrophies. For example, superficial variants and recurrent dystrophies after penetrating keratoplasty may lend themselves to treatment with excimer laser phototherapeutic keratectomy (PTK).
GRANULAR DYSTROPHY (GROENOUW TYPE I)
Clinical Features
Granular dystrophy, also called Groenouw type I dystrophy, is inherited as an autosomal-dominant dystrophy, with 100% penetrance (3, 4, 5, 6). The clinically obvious corneal lesions develop in early adolescence. These lesions are bilateral, symmetric, sharply demarcated grayish-white opacities of variable size and shape, or radial lines in the superficial stroma with intervening clear areas (Fig. 46-1 A,B). Opacities may vary in extent but rarely reach the limbus. Visual acuity is minimally affected in the early stages. Photophobia may occur due to scattering of light by the opacities. The opacities slowly progress, coalesce, and involve the deeper stroma. The overlying epithelium and the corneal sensations are intact.
Several variants of granular dystrophy are recognized. These include the progressive corneal dystrophy of Waardenburg and Jonkers (7) in which the onset is the first decade of life. Broad, oblique illumination on slit-lamp biomicroscopy offers the best visualization of the snowflakelike opacities. Retroillumination and indirect illumination reveal the fine granularity of a myriad punctate opacities. Visual acuity is severely affected by the fourth decade of life due to stromal opacification. Wittebol-Post and associates (8) have considered this dystrophy a variant of Reis-Bücklers’ dystrophy.
Rodrigues and colleagues (10) described another variant of granular dystrophy with onset in infancy. The deposits are superficial and cause painful recurrent erosions that require early surgical intervention. There are numerous deposits in the region of Bowman’s layer. The presence of classic granular dystrophy in parents or siblings may indicate that they are expressions of the same genotype (9,10).
In another variant of granular dystrophy, corneal opacities appear later in life. This alternative form of granular dystrophy is less severe and slower in progression than the other variants (8,11). The opacities have a breadcrumb-like appearance, and patients do not usually suffer from recurrent erosions.
Histopathology
The stroma contains hyaline eosinophilic deposits that stain intensely with Masson trichrome, weakly with periodic acid-Schiff (PAS) stain, and negatively with Verhoeff stain (Fig. 46-2). Congo red may stain the peripheral portions of the deposits (12,13). The epithelial basement membrane and Bowman’s layer may have area of thinning or may be absent in places (14).
Ultrastructurally, electron-dense rodlike deposits (100 to 500 μm wide) are present in the subepithelial region between the stromal lamellae and intracytoplasmically in keratocytes (9,15,16) (Fig. 46-3). The exact source of these deposits is unknown (17). Degenerative changes are
apparent in keratocytes, with cytoplasmic vacuolization and dilatation of endoplasmic reticulum (18). The deposits are either filamentous, homogeneous, or have a motheaten pattern (16,18). Tubular microfibrils can be seen surrounding these lesions. Histochemically, they contain tyrosine, tryptophan, and sulfur-containing amino acids (12). Moller and associates (19) demonstrated the presence of kappa and lambda light chains of immunoglobulin G in the lesions.
apparent in keratocytes, with cytoplasmic vacuolization and dilatation of endoplasmic reticulum (18). The deposits are either filamentous, homogeneous, or have a motheaten pattern (16,18). Tubular microfibrils can be seen surrounding these lesions. Histochemically, they contain tyrosine, tryptophan, and sulfur-containing amino acids (12). Moller and associates (19) demonstrated the presence of kappa and lambda light chains of immunoglobulin G in the lesions.
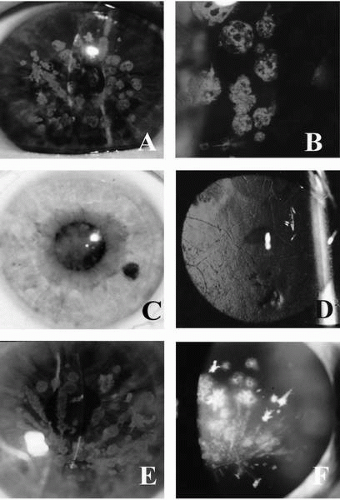 FIGURE 46-1. A,B: Granular dystrophy. Slit-lamp photograph demonstrates discrete, white, roundtooval, bread crumblike opacities with intervening clear areas that do not reach the limbus. C,D: Lattice dystrophy. Notice subepithelial opacity in the central cornea. Retroillumination shows branching, ropy lesions. E,F: Avellino dystrophy. (From Charukamnoetkanok P, Dohlman CH. Corneal dystrophies in the age of the human genome project. MEJO 2002;10:112-128, with permission.) |
Genetic Studies
Granular dystrophy (20) is one of the four dystrophies that have been mapped to the same chromosomal region on
5q31 (21). The other dystrophies are Reis-Bücklers corneal dystrophy (22), lattice corneal dystrophy type I (CDL1) (21), and Avellino corneal dystrophy (ACD) (23). Munier and associates (24) generated a YAC contig of the linked area, and following cDNA selection, recovered the BIG-H3 gene, which encodes the structural protein keratoepithelin. This observation established a common molecular origin of the four 5q31-linked corneal dystrophies (24).
5q31 (21). The other dystrophies are Reis-Bücklers corneal dystrophy (22), lattice corneal dystrophy type I (CDL1) (21), and Avellino corneal dystrophy (ACD) (23). Munier and associates (24) generated a YAC contig of the linked area, and following cDNA selection, recovered the BIG-H3 gene, which encodes the structural protein keratoepithelin. This observation established a common molecular origin of the four 5q31-linked corneal dystrophies (24).
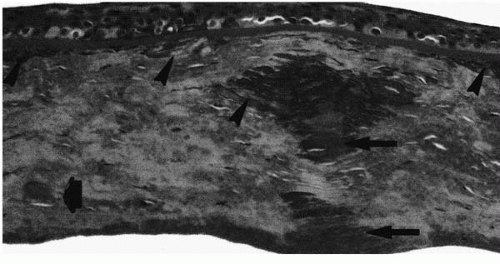 FIGURE 46-2. Granular dystrophy. Masson trichrome staining of a corneal button from a patient with granular dystrophy shows granular hyaline deposits at the level of superficial and midstroma. (From Santo RM, et al. Clinical and histopathologic features of corneal dystrophies in Japan. Ophthalmology 1995; 102:557-567, with permission.) |
Treatment
Intervening clear areas between the opacities allow unobstructed vision. Thus, patients are asymptomatic in the early stages. As the disease progresses, visual acuity is reduced and superficial keratectomy may be visually beneficial (25). In the superficial variant, treatment with the excimer laser (PTK) may be an attractive alternative to lamellar keratectomy. The goal of PTK is to remove the anterior corneal opacity and smooth the anterior refractive surface. PTK is performed after removal of the epithelium either mechanically or by ablation with the excimer laser. If the epithelial surface is rough, application of a smoothing or modulating agent may be necessary to create a more regular surface prior to laser ablation of the cornea.
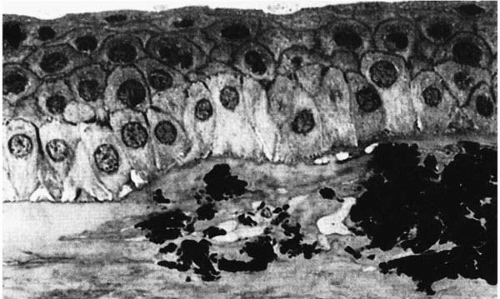 FIGURE 46-3. Semifine micrograph of the cornea from a patient with granular dystrophy shows deposits in the anterior stromal layer. (From Werner LP, et al. Confocal microscopy in Bowman and stromal corneal dystrophies. Ophthalmology 1999;106: 1697-1704, with permission.) |
Variable success rates ranging from 66% to 100% have been reported (26). Recurrences after keratectomy have been reported as early as 1 year (27,28) and are more frequent in the superficial variant. It is believed that in this disease, homozygous patients present earlier, have a more severe form, and have earlier recurrences following surgical intervention (6,29). Penetrating keratoplasty is required in the more advanced cases. Recurrences of granular dystrophy in the graft manifest as diffuse haze in the peripheral graft or as granular lesions in the stroma. The diffuse haze occurs due to growth of fibrous tissue between the epithelium and Bowman’s layer and it can be stripped by superficial keratectomy.
AVELLINO CORNEAL DYSTROPHY
In 1988, Folberg and associates (23) first reported three families with histologic features of both granular and lattice dystrophies (Figs. 46-4, 46-5 and 46-6). Because all of these patients traced their origins to Avellino, Italy, it was later named Avellino corneal dystrophy by Holland and colleagues (30).
Clinical Features
Avellino dystrophy is inherited as an autosomal-dominant trait with very high penetrance (greater than 90%) and moderately variable expressivity. Males and females are equally affected. The dystrophy usually appears in the first or second decade of life. Three clinical signs characterize Avellino corneal dystrophy: (a) discrete anterior stromal, gray-white granular deposits; (b) mid to posterior stromal lattice lesions; and (c) anterior stromal haze.
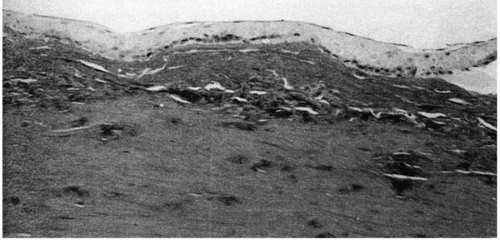 FIGURE 46-4. Avellino dystrophy. Masson trichrome stain of the patient with clinical finding of lattice dystrophy shows negative staining. (From Folberg R, et al. The relationship between granular, lattice type 1, and Avellino corneal dystrophies. A histopathologic study. Arch Ophthalmol 1994;112:1080-1085, with permission.) |
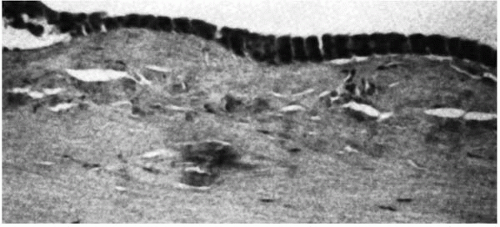 FIGURE 46-5. Avellino dystrophy. (From Folberg R, et al. The relationship between granular, lattice type 1, and Avellino corneal dystrophies. A histopathologic study. Arch Ophthalmol 1994; 112:1080-1085, with permission.) |
The earliest clinical evidence of this condition is discrete granular deposits, predominantly in the anterior third of the corneal stroma. Granules reach their mature size early and remain nearly stationary in size. They often coalesce to form linear opacities, especially in the inferior cornea. Lattice changes appear later in life and increase proportionally as the patient ages. They are located initially in the mid-and deep stroma and later involve the entire stroma (Fig. 46-1E,F). The lattice rods are thicker than those in lattice corneal dystrophy type I (LCD I) and bear a resemblance to lattice corneal dystrophy type IIIA (31). The last clinical sign to emerge is diffuse, ground-glass stromal haze located between the granular deposits.
Visual acuity is minimally affected, and depends on the predominant location of the deposits, which may be either central or peripheral. Patients complain of glare and decreased night vision. Recurrent erosions occur in the third and fourth decades of life, and the granular deposits that are superficial seem to be the cause of these erosions (32). Recurrent erosions are more common in patients with Avellino corneal dystrophy than in patients with typical granular dystrophy.
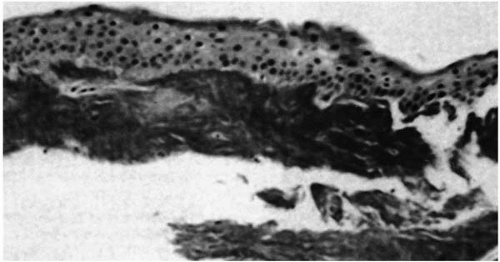 FIGURE 46-6. Avellino dystrophy. Superficial keratectomy specimen from a patient shows bright red granular deposit underneath the epithelium by Masson trichrome stain. (From Folberg R, et al. The relationship between granular, lattice type 1, and Avellino corneal dystrophies. A histopathologic study. Arch Ophthalmol 1994;112:1080-1085, with permission.) |
Histopathology
Granular deposits demonstrate both a hyaline appearance by hematoxylineosin staining and bright-red staining with Masson trichrome (Fig. 46-6). Numerous fusiform stromal deposits of amyloid that stain with Congo red and demonstrate birefringence and dichroism with the cross-polarization that is typical of lattice dystrophy are also present (30,33,34). The superficial granular deposits are ultrastructurally homogeneous and electron dense (Fig. 46-7). Microfilaments suggestive of amyloid can be seen in some of these deposits (Fig. 46-8).
Genetic Studies
Earlier reports had suggested that there may be a close relationship between lattice type I and granular dystrophy (12,25). Evidence illustrated that mutations in the BIG-H3 gene are the common molecular origin of the Avellino dystrophy, lattice type I dystrophy, granular dystrophy, and Reis-Bücklers dystrophy (24).
Treatment
Treatment is conservative and includes hypertonic saline and bandage contact lenses for recurrent erosions. PTK has been used to treat corneal erosions and to clear the central cornea of granular and lattice deposits (36). Treatment for decreased visual acuity also includes penetrating keratoplasty, which is generally required late in the course of the disease. Recurrences have been reported following keratoplasty as late as 9 years after the graft (30). The pattern
of recurrence seems to follow the natural history of the disease, with granular lesions being the first to appear.
of recurrence seems to follow the natural history of the disease, with granular lesions being the first to appear.
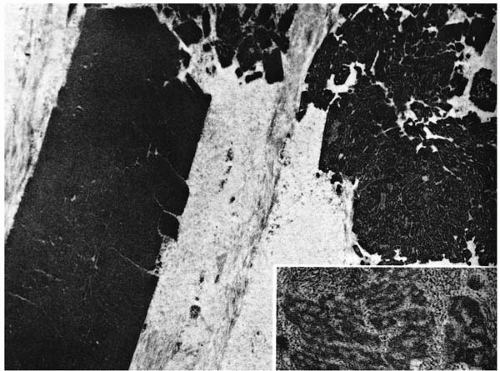 FIGURE 46-7. Avellino dystrophy. Electron micrograph shows electron dense deposits (left) and deposits that are apertured (right). Randomly oriented fibrils are located at the periphery of the apertured deposits (inset). (From Folberg R. Clinically atypical granular dystrophy with pathologic features of lattice-like amyloid deposits: study of these families. Ophthalmology 1988;95:46-51, with permission.) |
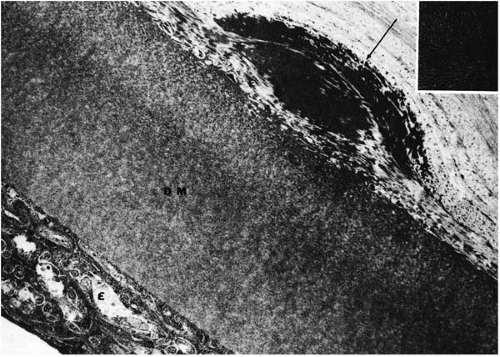 FIGURE 46-8. Avellino dystrophy. Electron micrograph demonstrates a fusiform deposit (arrow), composed of densely packed filaments (inset) is located anterior to Descemet’s membrane (DM). E = endothelium. (From Folberg R. Clinically atypical granular dystrophy with pathologic features of lattice-like amyloid deposits: study of these families. Ophthalmology 1988; 95:46-51, with permission.) |
LATTICE DYSTROPHY
There are four known types of lattice dystrophies: type I, Meretoja’s syndrome (type II), type III, and type IIIA.
Lattice Type I Corneal Dystrophy
Lattice dystrophy type I, also named Biber-Haab-Dimer dystrophy, was first described by Biber in the 1890s. The mode of inheritance is autosomal dominant with variable penetrance and expressivity. The disease may be detected in only a few members of the affected family. Other members may show only recurrent erosions with minimal stromal opacification (37).
Clinical Features
Lattice dystrophy appears in the first decade of life. Rarely does it appear after age 30 years (37,38). Corneal involvement is usually bilaterally symmetrical. However, unilateral cases have been reported in which the onset occurs later and the clinical course is less severe (39, 40, 41). In the early stages, the lesions appear as irregular lines and dots in the anterior axial stroma with clear intervening stroma or as a diffuse haze in the center. The faint central haze becomes denser with time, and it is this central disk-shaped opacity that eventually causes reduced visual acuity and may obscure the underlying lattice pattern (Fig. 46-1C). The dots and lines increase in size and number with time to become comma-shaped specks, thick ropy cords, and small nodules.
On direct illumination, gray opacities with irregular margins are seen, and on retroillumination, refractile rods with a clear core and double contour are obvious (Fig. 46-1D). The lines extend to the periphery, and go deeper in the stroma and toward the epithelium. As the opacities coalesce, a diffuse haze appears, involving the anterior and midstroma (Fig. 46-1C). At this stage the lattice dystrophy may be difficult to distinguish from granular or macular dystrophies, but careful examination usually reveals the typical branching lattice lines. The lattice filaments range from fine lines to coarse bands with nodular dilations. The lattice lines branch dichotomously near their central terminations and overlap one another at various stromal levels to create a lattice pattern. In advanced cases the lattice lines fluoresce under cobalt-blue slit-lamp illumination. Pseudofilaments may form due to the linear configuration of dots.
Epithelial erosions secondary to involvement of the subepithelial area and Bowman’s layer occur later in life and are the cause of discomfort and decreased visual acuity due to irregular astigmatism. They remain a serious problem until the third or fourth decade of life, after which they diminish in frequency. The corneal sensations decrease with concomitant symptomatic improvement. Vascularization rarely occurs.
An association between vestibulocochleopathy and lattice corneal dystrophy has been reported in a case of nonfamilial amyloidosis (42). Coexistence of progressive external ophthalmoplegia and corneal lattice dystrophy was seen in a family pedigree of 33 patients (43).
Lattice dystrophy should be differentiated from a nonprogressive, nonhereditary condition, polymorphic amyloid degeneration (parenchymatous dystrophy of Pillat) (44), in which stromal amyloid deposits are seen in patients above 60 years of age. Lattice lines can also be produced by hydroxyethylmethacrylate (HEMA) contact lenses but these disappear on removal of the contact lens.
Histopathology
Involvement of the epithelial or Bowman’s layer can be confirmed by polarization microscopy. Descemet’s membrane and the endothelium are unaffected. The deposits in lattice dystrophy are composed of amyloid, which has been confirmed by immunofluorescence (45), Congo red, PAS, and Masson’s trichrome staining. The deposits exhibit dichroism and manifest green birefringence (46,47). Dichroism is demonstrated by alternate red and green color when viewed through a rotating polarizing filter; and green birefringence is demonstrated by an intermittent yellowish-green against a black background when the lesion is viewed through two rotating polarizing filters. The deposits show a greenish-yellow fluorescence on staining with thioflavine T. Crystal violet staining shows metachromasia.
The amyloid protein is probably related to transthyretin (48,49).
The amyloid protein is probably related to transthyretin (48,49).
Ultrastructurally, the epithelium is of variable thickness, with degeneration of the basal epithelium. The basement membrane is thickened and continuous but without normal hemidesmosomes (50,51). Bowman’s membrane and superficial stroma contain amyloid structures, collagen fibrils, and fibroblasts. The stroma contains large irregular deposits that distort the normal configuration of the corneal lamellae. They contain highly aligned short, delicate, nonbranching electron-dense fibrils with diameters of 80 to 100 Å (52,53). There is a decrease in the number of keratocytes, with evidence of degeneration (46, 47, 48,54).
Amyloid is a complex of chondroitin, sulfuric acid, and protein found extracellularly. The amyloid seen in lattice dystrophy is distinct from amyloid in systemic amyloidosis or in primary or secondary amyloid degeneration of the cornea.
Genetic Studies
Linkage analysis and further studies revealed that the genetic mutation in patients with lattice dystrophy type I involves the BIG-H3 gene on chromosome 5q. Missense mutations occur at the CpG dinucleotide of two arginine codons: R124C in lattice dystrophy type I, R555W in granular dystrophy, R555Q in Reis-Bücklers’ dystrophy, and R124H in Avellino dystrophy (24).
Treatment
Managing lattice dystrophy in the early stages includes treatment of recurrent erosions. PTK is emerging as an attractive alternative to treat superficial corneal opacities and surface irregularities. The goal is to avoid or delay more invasive procedures such as lamellar and penetrating keratoplasty. PTK has been reported to be very successful for the treatment of lattice dystrophy (26). Penetrating keratoplasty is indicated when there is a decrease in visual acuity or, rarely, when the recurrent erosions become incapacitating. This is generally required in the third or fourth decade of life. Among major stromal dystrophies, recurrences after penetrating keratoplasty are more frequent in patients with lattice dystrophy. Recurrence of lattice dystrophy in the graft occurs as soon as 3 years postoperatively and is seen as dotlike filamentous subepithelial opacities or as a diffuse stromal haze (76).
Lattice Type II Corneal Dystrophy
Originally described by Meretoja (55,56) in the early 1970s, this systemic amyloidosis, also known as familial amyloid polyneuropathy type IV and amyloidosis V, is inherited as an autosomal-dominant disorder. The corneal changes appear in the third to fourth decade of life. Systemic involvement of the skin, peripheral nerves, and cranial nerves occurs later. This corneal dystrophy is less severe than is lattice type I dystrophy. Dots and lattice lines extend to the limbus and are fewer, thicker, and more radial. Erosions are less frequent and vision is unaffected until the seventh decade of life (57).
Histopathology
In type II lattice corneal dystrophy, amyloid deposits replace corneal nerves. Meretoja syndrome can be retrospectively diagnosed from corneal buttons based on the fact that monoclonal antibody to gelsolin labels almost all the deposits in contrast to the faint labeling of only some of the deposits in corneas from patients with type I lattice corneal dystrophy (58,59).
Genetic Studies
Lattice corneal dystrophy type II is caused by a specific mutation (asp187-to-asn) in the gelsolin gene on chromosome 9q34 (60). The amyloid protein found in patients with familial amyloidosis of the Finnish type (FAF) has been found to be an internal degradation fragment of gelsolin, with substitution of asparagine for aspartic acid at position 15, which is due to a guanine-to-adenine transversion in the gene. This mutation co-segregates with the disease phenotype and can be used as a diagnostic assay, including in prenatal evaluation (61).
Lattice Type III and Type IIIA Corneal Dystrophies
Reported in two families in Japan (62,63), type III lattice dystrophy manifests as thick translucent lattice lines and diffuse subepithelial opacities in the cornea. Lattice dystrophy type III may be autosomal recessive. The onset occurs after age 40, and vision is not affected until 70 years of age. Corneal erosions do not occur (31,62). A variant of this dystrophy (lattice type IIIA) has a similar clinical appearance. However, later age of onset (70 to 90 years), the occurrence of recurrent erosions, and an autosomal-dominant inheritance pattern differentiate it from lattice type III (64). Unilateral occurrence of type III lattice dystrophy has been reported in two cases and was confirmed by electron microscopy based on the presence of typical amyloid fibrils (65,66).
Histopathology
Both lattice dystrophy type I and type III deposits contain protein AA and protein AP (31,62,63) (Fig. 46-9). Fluorescent staining for immune deposits such as kappa chains, lambda chains, immunoglobulin C (IgC), IgM, and IgA are negative (67,68), unlike the findings in primary or secondary systemic amyloidosis in which these
proteins are present. The deposition of amyloid occurs either due to release of lysosomal enzymes by abnormal keratocytes (69,70) or by abnormal keratocyte synthesis (71,72). It is believed that sequestration of glycoprotein in the corneal stroma from plasma membranes of epithelial cells may stimulate amyloid deposition. Elastotic degeneration within amyloid deposits has been reported (73). Antibodies to C-reactive protein in the epithelium of corneas affected by lattice dystrophy has been reported, although the significance of this is uncertain (74).
proteins are present. The deposition of amyloid occurs either due to release of lysosomal enzymes by abnormal keratocytes (69,70) or by abnormal keratocyte synthesis (71,72). It is believed that sequestration of glycoprotein in the corneal stroma from plasma membranes of epithelial cells may stimulate amyloid deposition. Elastotic degeneration within amyloid deposits has been reported (73). Antibodies to C-reactive protein in the epithelium of corneas affected by lattice dystrophy has been reported, although the significance of this is uncertain (74).
 FIGURE 46-9. Comparison of lattice corneal dystrophy type III (left) and type I (right). Lattice dystrophy type III has uniform epithelium with a band of amyloid beneath intact Bowman’s layer, and had much larger amyloid stromal deposit than type I. (From Hida T, et al. Histopathologic and immunochemical features of lattice corneal dystrophy type III. Am J Ophthalmol 1987;104:249-254, with permission.) |
Genetic Studies
Yamamoto and colleagues (64) found that the mutation in type IIIA lattice corneal dystrophy was the missense change Pro501Thr in the keratoepithelin gene.
MACULAR DYSTROPHY (GROENOUW TYPE II)
Macular corneal dystrophy (MCD; also known as Groenouw type II) is divided into three subgroups on the basis of immunohistochemical studies (75, 76, 77, 78) and serum analysis of antigenic keratan sulfate (AgKS). In MCD type I, the more common subgroup, there is a virtual absence of AgKS in the serum and cornea. MCD type IA is differentiated from type I by the presence, in the former, of highly sulfated AgKS within corneal keratocytes in patients with undetectable amount of this form of AgKS in serum (78,79). In MCD type II, the AgKS is present in cornea and serum (75).
Clinical Features
Macular corneal dystrophy is transmitted as an autosomal-recessive trait. It is the least common of the three classic stromal dystrophies (granular, lattice, and macular), but it is the most severe. The corneal changes usually are first noted between 3 and 9 years of age, when a diffuse clouding is seen in the central superficial stroma (Fig. 46-10). Unlike granular dystrophy, the macular opacities have indistinct edges, and the intervening stroma is not clear. The clouding extends peripherally to the limbus and into the deeper stoma as the disease progresses.
By the teens, the opacification involves the entire thickness of the cornea and may extend out to the limbus. Within this sea of haziness are gray-white, denser opacities with
ill-defined borders. These denser, macular opacities can protrude anteriorly, resulting in irregularity of the epithelial surface, or posteriorly, causing irregularity, grayness, and guttate appearance of Descemet’s membrane. This may result in progressive loss of vision, irritation, and photophobia. These opacities can enlarge with time and coalesce. Corneal thinning, confirmed by central pachymetry, has been documented (80,81). In most cases, vision is severely impaired by age 20 or 30. Recurrent erosions are seen, but they are less frequent than in patients with lattice dystrophy. Heterozygous carriers do not manifest corneal abnormalities.
ill-defined borders. These denser, macular opacities can protrude anteriorly, resulting in irregularity of the epithelial surface, or posteriorly, causing irregularity, grayness, and guttate appearance of Descemet’s membrane. This may result in progressive loss of vision, irritation, and photophobia. These opacities can enlarge with time and coalesce. Corneal thinning, confirmed by central pachymetry, has been documented (80,81). In most cases, vision is severely impaired by age 20 or 30. Recurrent erosions are seen, but they are less frequent than in patients with lattice dystrophy. Heterozygous carriers do not manifest corneal abnormalities.
 FIGURE 46-10. A,B: Macular dystrophy. The opacities have indistinct edges, the intervening stroma is not clear, and the clouding extends peripherally to the limbus. (From Charukamnoetkanok P, Dohlman CH. Corneal dystrophies in the age of the human genome project. MEJO 2002; 10:112-128, with permission.) |
Biochemical studies have shown decreased α-galactosidase in keratocytes from cornea with macular dystrophy (82), and that the primary defect is in the synthetic pathway of the KS proteoglycans (82, 83, 84). The pathogenesis of macular dystrophy was thought to result from incomplete glycosaminoglycan sulfation (85). This view was supported by the fact that monoclonal antibodies demonstrated the absence of sulfated KS in the cornea and serum of patients with macular dystrophy (type I) (84). However, a new subtype of macular dystrophy was immunohistochemically identified in 1988, in which KS is present in the cornea and serum (type II) (75,76). Type I macular dystrophy is most prevalent in Europe and in North America. It is characterized by the absence of KS in the cornea as well as in the serum, and it may represent a more widespread systemic disorder of KS metabolism (86). Type II macular dystrophy may be more prevalent in Japan, based on a limited study by Santo and associates (87).
Histopathology
In macular dystrophy, the cornea synthesizes chondroitin/dermatan sulfate proteoglycans that are larger than normal and oversulfated (85). Histologically, macular dystrophy is characterized by the accumulation of glycosaminoglycans (52,88) (acid mucopolysaccharide) between the stromal lamellae, underneath the epithelium, within stromal keratocytes, and within the endothelial cells (52,88, 89, 90, 91). The glycosaminoglycans stain intensely with Alcian blue and with colloidal iron, minimally with PAS, and not at all with Masson’s trichrome. Birefringence is decreased. Proteoglycan-specific cuprolinic blue staining has revealed that these proteoglycans accumulate and aggregate in both type I and type II macular dystrophy corneas (Fig. 46-11). Degeneration of the basal epithelial cells and focal epithelial thinning is seen over the accumulated
material (92). Bowman’s layer is thinned or absent in some areas. The accumulated material varies; its staining by different anti-KS antibodies varies among patients (75,76).
material (92). Bowman’s layer is thinned or absent in some areas. The accumulated material varies; its staining by different anti-KS antibodies varies among patients (75,76).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


