glands, meibomian gland, conjunctiva, and cornea, suggesting that androgens influence their structural organization and functional activity, and have an especially profound impact on the immunology, molecular biology, and secretory capacity of the lacrimal gland (9). Such proven androgen effects have led to the idea that androgen deficiency states such as SS, SLE, RAs, aging, and the use of antiaging medications may be very important in the pathogenesis of dry eyes (10). The androgen binding to the receptors in the acinar nuclei of the exocrine glands (the lacrimal gland in the context of xerophthalmia) leads to an altered expression of numerous cytokines and proto-oncogenes. The activity of androgens in the exocrine glands is thought to induce the accumulation of antiinflammatory cytokines such as transforming growth factor-β (TGF-β) (10). The reduction of the androgen level below a certain threshold may result in the release of proinflammatory cytokines such as interleukin-1β (IL-1ß), IL-2, interferon-γ, and tumor necrosis factor-α (TNF-α) by the lymphocytes entering the gland (11). Experimental evidence has shown that progressive CD4+ T-cell and B-cell infiltration occurs in lacrimal and salivary glands of patients with SS (12). Alterations of the nerve fibers among the glandular acini are superimposed on the inflammatory process in these patients. It is also possible that the central nervous system plays an important role in the cascade of events that occur in SS where the amount of tear flow is initially decreased through cellular infiltration of the tear gland. Increased friction between the lids dry cornea may cause exfoliation of the superficial corneal epithelium, resulting in constant repeated C-fiber nerve stimulation, which becomes dominant in time and acts in combination with central parasympathetic inhibition of tear flow. This could also explain the clinical and experimental discrepancies of the concept of infiltration and destruction of the lacrimal gland as a single cause of tear flow depression. In addition, central neural mechanisms may also explain those cases in which patients have subjective rather than objective improvement of complaints (13).
TABLE 29-1. EVOLUTION OF HISTORICAL ISSUES RELATED TO SJÖGREN’S SYNDROME | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Numerous investigators have attempted to analyze the association of primary SS with cytokine polymorphisms. Both human and animal studies indicate the involvement of IL-10 in the pathogenesis of primary SS, and mice transgenic for IL-10 develop a Fas-ligand mediated exocrinopathy that resembles SS (17,18). A recent study described an association between primary SS and IL-10 promoter polymorphisms in a cohort of Finnish individuals, and a specific haplotype was found to correlate with high plasma levels of IL-10 (19). Conversely, no association was found for IL-10 promoter polymorphism and primary SS or the presence of Ro autoantibodies in an Australian cohort of primary SS patients (20). The IL-1 receptor antagonist regulates IL-1 activity in inflammatory disorders by binding to the IL-1 receptor and thus, inhibiting its activity. The human IL-1 receptor antagonist gene (IL1RN) has a variable allelic polymorphism within intron 2. An increased frequency and carriage rate of the ILRN*2 allele has been found in primary SS (21,22).
TABLE 29-2. CLINICAL SPECTRUM OF SJÖGREN’S SYNDROME | |||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
(30). In other words, the infiltration of lymphocytes alone did not cause glandular dysfunction. Apoptosis of acinar cells may explain the differences.
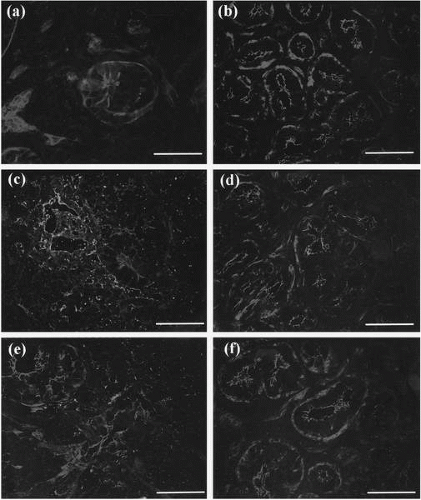 FIGURE 29-1. Quantitative evaluation and evidence of apoptotic mechanisms in Sjögren’s syndrome (SS): double staining of actin and Apo2.7, Fas, or FasL in lacrimal gland. A,C,E: Lacrimal glands from SS. B,D,F: Lacrimal glands from non-SS were stained with Apo2.7 (A,B), Fas (C,D), and FasL (E,F) antibody followed by rhodamine-phalloidin. Bars: 50 μm. A: SS lacrimal gland. Note the (+) acinar cells. B: Non-SS lacrimal gland. No acinar cells are stained with Apo2.7 antibody. C: SS lacrimal gland. Note the (+) acinar cells, D: Non-SS lacrimal gland. Some acinar cells are stained with Fas antibody. E: SS lacrimal gland. Note (+) staining of infiltrating lymphocytes. F: Non-SS lacrimal gland. FasL antibody did not stain any cells. (From Tsubota K, Fujita H, Tsuzaka K, et al. Quantitative analysis of lacrimal gland function, apoptotic figures, Fas and Fas ligand expression of lacrimal glands in dry eye patients. Exp Eye Res 2003; 76:233-240. Copyright 2003, with permission from Elsevier.)(see color image) |
disequilibrium, resulting in difficulties in establishing which of the genes contains the locus that confers the risk (52). It has been claimed that SS patients with DQ1/DQ2 alleles have a more severe autoimmune disease than do patients with any other allelic combination at HLA-DQ, and the DR3-DQ2 haplotype has been indicated as a possible marker for a more active immune response in Finnish patients with SS (53). Autoantibodies to Ro/SSA and La/SSB have been found to be associated with DR3, DQA, and DQB alleles (54, 55, 56). In Japan, HLA class II allele association has been reported to differ among anti-Ro/SSA (+) individuals according to the presence or absence of coexisting anti-La/SSB (8). The contribution of Ro/SSA and La/SSB in SS is not fully understood. It is not known how tolerance breakdown and autoantibody response to Ro/SSA and La/SSB is generated. The ribonucleoproteins are endogenous proteins that are normally hidden from the immune system and should not give rise to abnormal B-cell responses. However, stress, such as ultraviolet radiation, viral infections, and apoptosis have been suggested to lead to cell surface exposure of previously hidden autoantigens to the immune system (57). Genes that encode transporters associated with antigen processing (TAP genes) have also been associated with susceptibility to SS (58). Others have indicated a putative role for the cysteine-rich secretory protein 3 (CRISP-3) gene as an early response gene that may participate in the pathophysiology of the autoimmune disease in SS (59).
apical membranes were normally distributed in all patients (Table 29-3). In a recent study, we found that AQP5 increased in the tears of patients with severe SS compared with control subjects (64). This finding corresponds to a previous report that leakage of AQP5 in the tear was related to the lacrimal gland damage in experimental dacryoadenitis models. Although significant correlations exist between tear lactoferrin and EGF and clinical indices such as tear function index, Schirmer test and rose-Bengal staining scores, no correlation between AQP5 levels and clinical indices was observed. These differences suggest that lactoferrin and EGF produced by the lacrimal gland directly preserve ocular surface function, whereas AQP5 has not a direct but rather a secondary influence on the ocular surface disorder, caused by a decrease in the number of lacrimal gland cells (64).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


