Scleral Transplantation
William J. Power
Andra S. Bobart
The sclera has been successfully used for transplantation in dental, nasal, plastic-reconstructive, and ocular surgeries. Sclera may be transplanted for scleral ectasia, staphyloma, scleral perforation, glaucoma implant surgery, and patch graft repair of leaking filtering blebs (1).
Scleral tissue loss occurs secondary to inflammatory, necrotizing, traumatic, infective, iatrogenic, or idiopathic mechanisms. Recurrent scleritis leads to scleral thinning, which may eventually result in uveal prolapse and full-thickness scleral perforation. This tissue loss can be reinforced, repaired, or replaced with human sclera.
HISTORY
Historically, enucleation was performed in eyes with very severe uveal ectasia to avoid the risk of imminent rupture of the globe. Many materials have been used for scleral repair, including fascia lata, auricular cartilage, periosteum, autologous sclera, and preserved sclera (2, 3, 4, 5, 6). The use of autologous donor sclera was described at least as early as 1948 by Larsson for repair of a corneal perforation, and in 1953 homologous donor sclera was used by Paufique for the repair of scleromalacia perforans (5,6). It has been suggested by Curtin et al. that the use of homologous sclera may be better than autologous sclera in cases of scleromalacia perforans because the entire body collagen may be affected by the rheumatoid disease (7). The use of autologous sclera has largely been replaced with preserved human sclera from eye bank donor eyes. The use of sclera from an older donor may be preferable because it has greater rigidity and higher tensile strength.
HISTOLOGY AND IMMUNOLOGY
The sclera is relatively avascular, with blood vessels traversing it rather than supplying it directly. It is also relatively acellular, with up to 75% of its dry weight being collagen and up to 70% of its intact weight being water. The remainder of the sclera is made up of noncollagenous protein, proteoglycans, and mucopolysaccharides (8, 9, 10). Although collagen has been shown to induce specific humoral and cell-mediated immune reactions in experimental animals, the sclera itself has been repeatedly shown to have low antigenicity. Iacono et al. confirmed this in 1980 after testing the reactivity of human peripheral blood leukocytes to extracts of sclera (11, 12, 13, 14, 15). Scleral homograft rejection, therefore, is rarely encountered.
Necrotizing scleritis is the main cause of extreme scleral thinning requiring scleral transplantation. Histopathologic studies on patients with necrotizing scleritis demonstrate a mixed mononuclear and neutrophil infiltrate and fibrinoid necrosis of vessel walls. Immune complex deposition and a perivasculitis may also be seen (16).
Two main types of histopathosis have been described in necrotizing scleritis (17). The first is zonal necrotizing granulomatous inflammation, which surrounds a central necrotic area and is associated with vasculitis. This is mainly associated with rheumatoid arthritis, relapsing polychondritis, Goodpasture’s syndrome, Wegener’s granulomatosis, collagen vascular disease, and previous herpes zoster ophthalmicus. The second main type is necrotizing inflammation with multiple microabscesses. This may be associated with either positive or negative cultures for microorganisms. Proteoglycans are absent in areas of active necrotizing scleritis, even though the collagen fibers themselves are initially intact. This may be due to proteoglycan resorption by the products of inflammatory cells, by cytokines, and by active resident fibroblastic cells. The latter are most likely largely responsible for the extracellular degradation of collagen (18,19). Matrix metalloproteinase-3 (stromelysin) has been identified as one of the collagendegrading enzymes in scleritis (20). A combination of the proinflammatory cytokines tumor necrosis factor-α and interleukin-1α may be responsible for enhanced matrix metalloproteinase-3 expression, thereby leading to increased tissue destruction (21). After scleral allografting, there is a severe immune reaction of edema, hyperemia, and a cellular infiltrate around the graft. After approximately 20 days, there is a reduction in this immune reaction that coincides with the extraction of glycosaminoglycans from
the collagen fibers of the allograft. It has been shown that the partially extracted glycosaminoglycans are gradually replaced by the recipient tissue, and a no-scar regenerate develops. There is fourfold less scarring where there is at least a 70% structural similarity between the donor and host tissue (13).
the collagen fibers of the allograft. It has been shown that the partially extracted glycosaminoglycans are gradually replaced by the recipient tissue, and a no-scar regenerate develops. There is fourfold less scarring where there is at least a 70% structural similarity between the donor and host tissue (13).
CAUSES OF SCLERAL TISSUE LOSS
The sclera was described by Duke-Elder as being “inert and purely supportive in function” (22). Loss of this support due to progressive collagen destruction and thinning can cause the underlying choroid to bulge and cause scleral ectasia. The uvea may even break through the overlying thin covering of sclera and the conjunctiva. Thin sclera is blue in color, and with progressive thinning the affected area becomes dark because of the color of the underlying uvea.
Scleral thinning may be inherited in conditions such as in Marfan’s syndrome, osteogenesis imperfecta, or Ehlers-Danlos syndrome (23, 24, 25). It may also be acquired in conditions such as pathologic myopia, iron-deficiency anemia, perilimbic scleromalacia, and after surgery (26, 27, 28, 29, 30, 31). In staphyloma secondary to glaucoma, Thale et al. demonstrated a loss of the normal rhombic collagenous alignment of the inner sclera with scleral protrusion between parallel collagen bundles (8). And although scleritis rarely leads to frank perforation of the globe, postsurgical scleritis and scleral inflammation secondary to granulomatous, nongranulomatous, infective, and vasculitic disease have all been shown to lead to extreme scleral thinning, ectasia, and subsequent perforation.
Necrotizing scleritis is characterized as either necrotizing scleritis with inflammation (Fig. 55-1) or necrotizing scleritis without inflammation (scleromalacia perforans; Fig. 55-2). Necrotizing scleritis is frequently associated with the development of peripheral keratopathy (Fig. 55-3). The presence of scleritis with peripheral ulcerative keratopathy has a poor prognosis (32). Both types of necrotizing scleritis can lead to spontaneous or traumatic perforation and are associated with rheumatoid arthritis and with other autoimmune or systemic diseases. In one case series described by Sainz de la Maza et al., approximately 95% of patients with necrotizing scleritis had an associated systemic disease. Necrotizing scleritis was also seen in this study to be the most common type of scleritis when presenting as the first manifestation of systemic disease (33). Rheumatoid arthritis is the disease most commonly associated with scleromalacia perforans and with bilateral scleritis. Wegener’s granulomatosis is also associated with necrotizing scleritis with inflammation. Other associated systemic diseases include relapsing polychondritis, polyarteritis nodosa, and inflammatory bowel disease. Necrotizing scleritis is uncommon in patients with juvenile idiopathic arthritis, Behâet’s disease, the spondyloarthropathies, and systemic lupus erythematosus. Up to 10% of patients with necrotizing scleritis require some form of surgical intervention (32,33). Watson and Hayreh reported systemic or ocular complications in 60% of their patients with necrotizing scleritis and a mortality rate of 29% within 5 years (34). The presence of vasculitis with necrotizing scleritis can be associated with significant systemic morbidity (35).
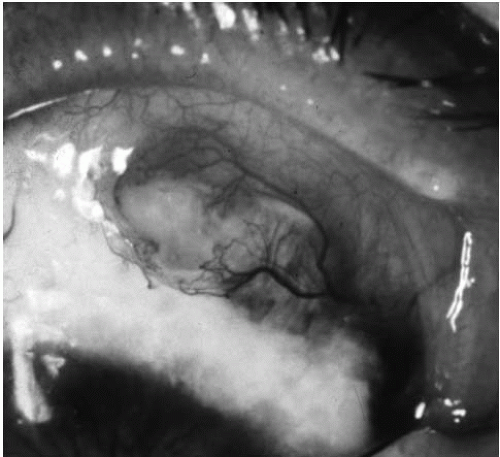 FIGURE 55-1. Necrotizing scleritis with inflammation.(see color image)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|