Purpose
To assess the safety, tolerability, pharmacokinetics, and dose-limiting toxicity of single intravitreal injection of Sirna-027, a small interfering RNA targeting vascular endothelial growth factor receptor-1, in patients with choroidal neovascularization (CNV) resulting from neovascular age-related macular degeneration (AMD). Secondary objectives included assessment of anatomic changes in retinal thickness, size of CNV, and changes in visual acuity.
Design
Prospective, open-label, single-dose, dose-escalation phase 1 study.
Methods
Twenty-six eyes of 26 patients with a median age of 82 years and CNV resulting from AMD who had previous treatments with other therapies were treated at 2 academic retinal practices. Patients received a single dose of Sirna-027 (100, 200, 400, 800, 1200, or 1600 μg/eye). Blood was sampled for pharmacokinetic analysis at 1, 4, and 24 hours after injection and on day 7. Patients underwent ophthalmic examinations including visual acuity, fluorescein angiography, and optical coherence tomography at screening and days 7, 14, 28, and 84. The main outcome measures were adverse reactions and dose-limiting toxicities.
Results
Intravitreal injection of a single dose of Sirna-027 from 100 to 1600 μg was well tolerated in patients with AMD, with no dose-limiting toxicity found. Adverse events were mild to moderate in severity. Adjusted mean foveal thickness decreased within 2 weeks after study treatment. The decrease was most pronounced in the 100- and 200-μg doses.
Conclusions
A single intravitreal dose of Sirna-027 up to 1600 μg/eye was well tolerated in patients with CNV resulting from neovascular AMD that had been refractory to other therapies. Stabilization or improvement in visual acuity and foveal thickness was observed. No dose-response or dose-limiting effects were noted.
Age-related macular degeneration (AMD) is the leading cause of irreversible vision loss in the Western world. Since the publication of the Minimally Classic/Occult Trial of the Anti-VEGF Antibody Ranibizumab in the Treatment of Neovascular AMD (MARINA) and Anti-VEGF Antibody for the Treatment of Predominantly Classic Choroidal Neovascularization in AMD (ANCHOR) trials, demonstrating clinical treatment effects greater than had been seen hitherto, inhibition of vascular endothelial growth factor (VEGF) A has become the standard of care for most presentations of neovascular AMD. VEGF has 2 cell surface receptors, both of which are tyrosine kinases: VEGF receptor 1 (VEGFR-1) and VEGF receptor 2 (VEGFR-2). These receptors are potential targets for treatment of neovascular AMD, not only because VEGF inhibition is so successful, but also because inhibition of individual receptors may allow particular aspects of the angiogenic process to be modulated, and inhibition of a receptor with multiple ligands may have a greater effect than simply inhibiting the action of a single ligand.
VEGFR-1 is the receptor for both vascular endothelial growth factor B and placental growth factor, both of which may play a role in pathologic neovascularization. VEGFR-2 is the major mediator of the mitogenic, angiogenic, and permeability-enhancing effects of VEGF. The functions of VEGFR-1 are very complex, with both positive and negative angiogenic effects. It is upregulated by hypoxia and plays a role in hematopoiesis, recruitment of monocytes, induction of matrix metalloproteinases, and release of growth factors from endothelial cells. It also may play an inhibitory role by sequestering VEGF and preventing its interaction with VEGFR-2. Activation of VEGFR-1 by placental growth factor results in transphosphorylation of VEGFR-2, thus amplifying angiogenesis through VEGFR-2.
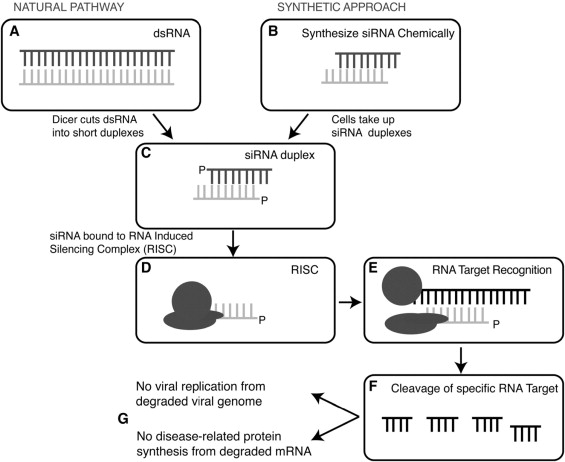
Sirna-027 (also known as AGN 211745) is a chemically modified small interfering RNA (siRNA) molecule that targets a conserved region of human, cynomolgus monkey, mouse, and rat VEGFR-1 mRNA molecules. Sirna-027 reduces pathologic neovascularization in a mouse laser-induced choroidal neovascularization (CNV) model in mouse oxygen-induced retinopathy. siRNA molecules induce gene silencing by binding to complementary target RNA molecules in association with the nucleolytic cytoplasmic protein complex known as the RNA-induced silencing complex ( Figure 1 ).
Nonclinical safety studies were conducted and included a single-dose intravitreal pharmacokinetic study in rabbits, single-dose intravitreal toxicity studies in rats and cynomolgus monkeys, and a single-dose intravenous toxicity study in rats (data on file). The primary effect of Sirna-027 in rats and monkeys was mild ocular inflammation detected by ophthalmoscopy, histopathologic analysis, or both at the highest doses, which was reversible and did not progress with repeated dosing. The observed adverse effect levels defined in the nonclinical ocular safety studies were 9- to 12-fold times the starting dose in the current study.
Based on the preclinical activity and tolerability of Sirna-027, a phase 1 study was conducted. The study was designed to demonstrate the safety and tolerability of single ascending intravitreal doses of Sirna-027 and to evaluate the proof-of-principle biological activity of Sirna-027 in patients with CNV secondary to AMD.
Methods
Study Design
A prospective, open-label, single-treatment, dose-escalation phase 1 study was performed to assess the safety, tolerability, and potential biological activity of Sirna-027 in patients with CNV secondary to neovascular AMD. Patients were enrolled at 2 United States sites: the Cole Eye Institute (9 patients) and the Wilmer Eye Institute (17 patients).
The primary objectives of the trial were to assess the safety, tolerability, and dose-limiting toxicity (DLT) of a single dose of Sirna-027 when administered by intravitreal injection; to assess the plasma concentrations after a single intravitreal injection; and to determine the range of doses appropriate for the use in further clinical trials. Secondary objectives included assessment of changes in visual acuity (VA) and of anatomic changes in retinal thickening on optical coherence tomography (OCT), and lesion size and leakage on fluorescein angiography (FA).
Key Inclusion Criteria
Patients were eligible for inclusion if they were 50 years of age or older and had active, subfoveal CNV of any combination of occult and classic components secondary to AMD confirmed by fundus biomicroscopy and FA and otherwise met the following criteria: (1) best-corrected visual acuity in the study eye of 20/100 or worse but not worse than 20/800 on the Early Treatment Diabetic Retinopathy Study chart at 4 m; (2) clear ocular media and papillary dilatation to at least 4 mm with the use of standard mydriatics; (3) central subfield thickness of at least 250 μm as measured by time-domain OCT; (4) the neovascular lesion had to be composed of less than 50% blood and less than 50% scar and had a size of less than 12 Macular Photocoagulation Study disc areas.
At the time when the study was conducted, standard therapy for neovascular AMD included photocoagulation and photodynamic therapy. Antagonists of VEGF were not available outside of a clinical trial. All patients who were enrolled in the study had evidence of disease activity based on FA and OCT results, despite prior treatment with photodynamic therapy or other treatments. The patients had declined further treatment with photodynamic therapy. As the study proceeded, some phase 3 studies of VEGF antagonists also began. However, because many patients in this phase 1 Sirna-027 study had advanced disease with large lesion sizes, they were not eligible for these studies with VEGF inhibitors.
Key Exclusion Criteria
Patients were excluded if they (1) were women of child-bearing potential; (2) had any ocular condition that could affect vision or safety evaluation; (3) had any ocular condition, apart from AMD, that is associated with CNV, including pathologic myopia, defined as spherical equivalent of −8 diopters or more, presumed ocular histoplasmosis syndrome, angioid streaks, choroidal rupture, or multifocal choroiditis; (4) had undergone any intraocular surgery or retinal treatment with verteporfin photodynamic therapy (Visudyne; Novartis AG, Basel, Switzerland), or other standard or experimental AMD treatments (with the exception of vitamin and mineral supplementation) within 3 months of study entry; or (5) had been diagnosed with any of the following underlying systemic diseases: uncontrolled diabetes mellitus or presence of diabetic retinopathy, cardiac disease including myocardial infarction within 12 months before study entry, coronary disease associated with clinical symptoms, stroke (within 12 months of study entry), active bleeding disorders, any major surgical procedure within 1 month of study entry, and active peptic ulcer disease with bleeding within 6 months of study entry, or had received concomitant systemic therapy with corticosteroids (e.g., oral prednisone) or other anti-angiogenic drugs (e.g., thalidomide).
Treatment
Within 2 weeks of screening, eligible patients assigned to the first cohort of 3 patients received a single intravitreal injection of Sirna-027 in the study eye at a starting dose of 100 μg. Subsequent cohorts (3 to 6 patients) were treated with doses of 200, 400, 800, 1200, and 1600 μg/eye after review of the day 14 safety data from the previous cohort. In the absence of a DLT event being reported, dose escalation occurred. DLT was defined as any toxicity determined by the investigator to be related to the study drug and of such intensity or severity to preclude dose escalation. Systemic DLTs included severe or life-threatening toxicities or any significant toxicity deemed by the investigator to have been related to the study drug. Local anesthesia and topical antimicrobials were administered before study drug injection. The study dose of Sirna-027 was delivered at the appropriate concentration in a 100 μL volume via a 30-gauge needle, 3.5 to 4.0 mm posterior to the limbus.
Blood sampling for pharmacokinetic assessment was performed before injection and at 1, 4, and 24 hours after injection as well as on day 7. Patients also underwent complete ophthalmic examinations including VA measurements, slit-lamp biomicroscopy, applanation tonometry, FA, and OCT at screening and on days 7, 14, 28, and 84 (or early termination). Protocol best-corrected VA testing, slit-lamp biomicroscopy, and applanation tonometry also were performed on days 2 and 56, and OCT was carried out on day 56. Vital signs, toxicity, and concomitant medications were monitored at all posttreatment visits through day 84 (week 12). Patients underwent routine eye examinations at 6, 9, 12, 18, and 24 months after study treatment, which included fundus photography at all visits except for that at month 9.
Reading Center
The Retinal Imaging Research and Reading Center (RIRRC) at the Wilmer Eye Institute served as the reading center for FA and OCT analyses. All images were evaluated by 3 graders (R.C.A.S., S.M.S., H.T.) who were masked with respect to treatment dose.
Optical Coherence Tomography
OCT was performed using StratusOCT (Carl Zeiss Meditec, Dublin, California, USA).
The RIRRC provided clinical sites with detailed instruction for OCT image acquisition, and a representative from the RIRRC visited each study site to certify competence and compliance. Two standard protocols (6-mm fast macular thickness map and 6-mm cross-hair) and 1 modified acquisition protocol (3-line 8-mm papillomacular axis scan) were used. The 3-line, 8-mm papillomacular axis scans used the disc as a landmark to ensure reproducible placement of scan lines at each visit; one line was at the superior margin, one was at the inferior margin, and one passed through the center of the disc. By using a univariate landmark such as the optic disc, patients who could not fixate could be reproducibly scanned. The 6-mm linear cross-hair pattern was a high-resolution scan used to follow morphologic changes in the macula. The fast macular thickness map used 6 linear scans 6 mm in length centered on the patient’s fixation at equally spaced angular orientations. Retinal thickness at any point was defined as the distance between outer and inner hyperreflective bands of the OCT cross section. Foveal thickness (in micrometers; defined as the mean height of the neurosensory retina in a central 1-mm diameter area) and total macular volume (in cubic millimeters) were computed automatically by the StratusOCT software (version 4.0). Interpretation of OCT images is more difficult in patients with CNV than in patients with other diseases such as diabetic macular edema. In patients with CNV, the computer often misinterprets the inner and outer retinal borders, and therefore, the computer-generated retinal thickness measurements may be incorrect. Because of the advanced nature of the disease and extensive change in the RPE morphologic features, manual scan profiling was used to correct any artifacts produced by the automated analysis algorithm.
Fluorescein Angiography
A modified FA acquisition protocol was used for high-resolution image acquisition, and compliance was monitored by a site visit. Digital images were sent to the RIRRC and analyzed using EyeRoute Proview software (version 6.1; Anka Systems Inc, McLean, Virginia, USA). Two independent investigators (R.C.A.S., H.T.) graded each FA image for markers of disease activity (progression/regression), including blood and pigment epithelial detachment. Each FA image was analyzed further by manual tracing of lesion perimeter to determine the lesion size, location, composition, and maximum area and extent of leakage.
Statistical Analysis
Because this was a phase 1 safety study, there were no clinical efficacy end points in this trial. All patients who received any amount of study drug were included in the safety analysis. Visual acuity results, as measured on the Early Treatment Diabetic Retinopathy Study visual acuity chart at 4 m, were presented using descriptive statistics at baseline and each postbaseline time point. The final analysis was performed after all patients completed the day 84 study visit or an early termination visit. The Jonckheere-Terpstra exact test (2-sided) from a Monte-Carlo simulation was used to assess the evidence for a dose-related trend in change (number of letters and number of lines).
Anatomic outcomes as measured by changes in FA, fundus photography, and OCT evaluations were evaluated by masked graders at the RIRRC. The absolute and percent changes from baseline in area of CNV determined from FA images were summarized by dose level and time point. The Jonckheere-Terpstra exact test (2-sided) was used to assess the evidence for a dose-related trend in change from baseline. The absolute and percent changes from baseline in foveal thickness and macular volume, as measured by OCT, were presented by dose group and time point. The Jonckheere-Terpstra exact test (2-sided) was used to assess the evidence for a dose-related trend in the change for automated measurements of foveal thickness. The Jonckheere-Terpstra exact test (2-sided) from a Monte Carlo simulation was used to assess the evidence for a dose-related trend in change for adjusted measurements of foveal thickness, parafoveal thickness, and macular volume.
Results
Twenty-six patients with CNV secondary to AMD were enrolled and received treatment in this clinical trial. All 26 patients completed the study through day 84. Baseline demographic characteristics are summarized in the Table . Twenty of the 26 patients (76.9%) in this study were females, and all patients were white. The mean age for all patients was 81.5 years (range, 67 to 93 years). Eighteen of the 26 patients had received 1 or more prior treatments for AMD in the study eye. At baseline, the CNV type in the study eye was classified as predominantly classic in 10 patients (38.5%), minimally classic in 12 patients (46.2%), and occult in 4 patients (15.4%).
| 100 μg | 200 μg | 400 μg | 800 μg | 1200 μg | 1600 μg | Total | |
|---|---|---|---|---|---|---|---|
| Age (yrs) at baseline | |||||||
| No. | 4 | 3 | 3 | 6 | 6 | 4 | 26 |
| Mean (SD) | 84.5 (6.24) | 74.7 (7.51) | 86.0 (1.73) | 78.5 (5.50) | 83.0 (2.53) | 82.5 (2.89) | 81.5 (5.49) |
| Median | 83.5 | 75 | 85 | 78.5 | 83 | 82.5 | 82 |
| Range | 78 to 93 | 67 to 82 | 85 to 88 | 70 to 86 | 80 to 87 | 80 to 85 | 67 to 93 |
| Sex | |||||||
| Male | 1 (25.0%) | 2 (66.7%) | 1 (33.3%) | 1 (16.7%) | 1 (16.7%) | 0 | 6 (23.1%) |
| Female | 3 (75.0%) | 1 (33.3%) | 2 (66.7%) | 5 (83.3%) | 5 (83.3%) | 4 (100%) | 20 (76.9%) |
| Race | |||||||
| Asian | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Black | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| White | 4 (100%) | 3 (100%) | 3 (100%) | 6 (100%) | 6 (100%) | 4 (100%) | 26 (100%) |
| Hispanic | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Other | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Treatment history | |||||||
| Photodynamic therapy with verteporfin | 2 (50.0%) | 2 (66.7%) | 1 (33.3%) | 2 (33.3%) | 1 (16.7%) | 1 (25.0%) | 9 (34.6%) |
| Subfoveal laser photocoagulation | 2 (50.0%) | 0 | 0 | 0 | 1 (16.7%) | 0 | 3 (11.5%) |
| Intravitreal injection with triamcinolone | 1 (25.0%) | 2 (66.7%) | 0 | 0 | 0 | 0 | 3 (11.5%) |
| Other treatments | |||||||
| Clinical trial with adenoviral vector and pigment epithelial derived factor | 0 | 0 | 3 (100%) | 3 (50%) | 2 (33.3%) | 2 (50.0%) | 10 (38.5%) |
| Clinical trial with combretastatin infusions | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Injection of gas for pneumatic air displacement of subretinal hemorrhage | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| Laser retinopexy | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Pegaptanib (Macugen) | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Posterior sub-Tenon injection with triamcinolone | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| Rheopheresis | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| Clinical trial with VEGF-Trap infusions | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


