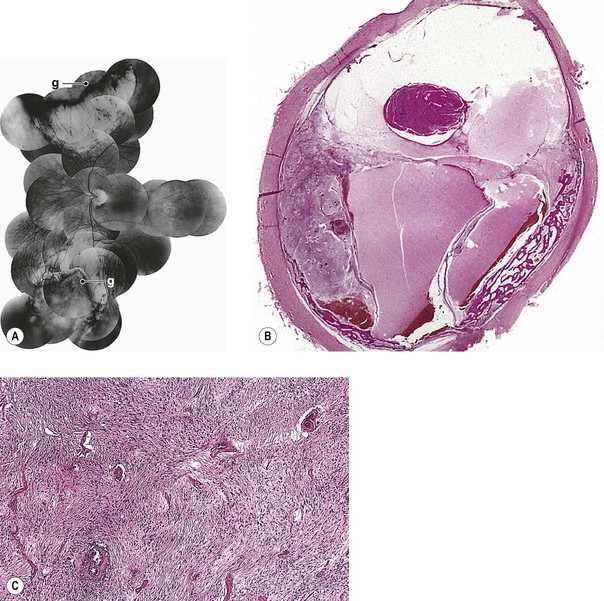
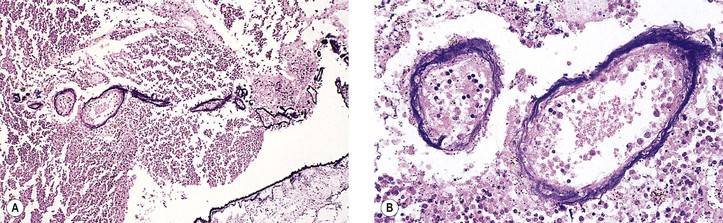
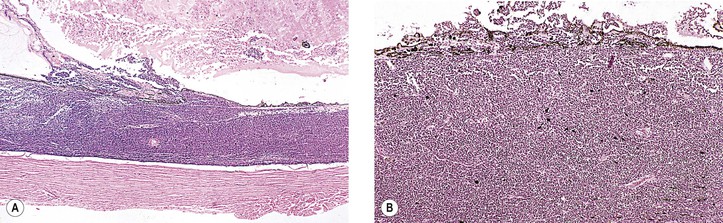
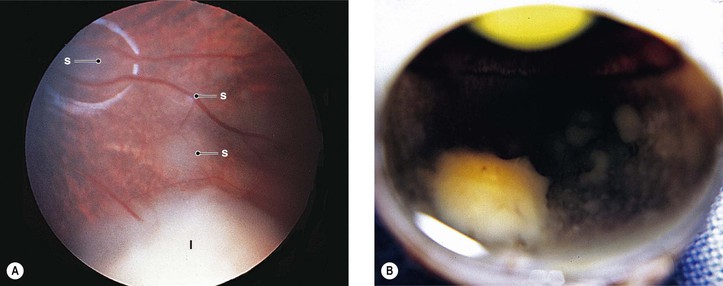
III. The incidence is approximately one in 18,000 live births in the United States, with a trend toward a higher prevalence than historically found (because of increased survival rate). B. The two clinical classification systems most commonly used for retinoblastoma are the Reese–Ellsworth classification (REC) (Table 18.1) and the International Classification (IC) (Table 18.2). TABLE 18.1 Reese–Ellsworth Classification for Intraocular Tumors A. Solitary tumor, smaller than 4 disc diameters (DD), at or behind the equator B. Multiple tumors, none larger than 4 DD, all at or behind the equator sight A. Massive tumors involving more than one half of the retina (Modified from National Cancer Institute: Retinoblastoma classification. Available at http://www.cancer.gov/cancertopics/pdq/treatment/retinoblastoma/HealthProfessional/page3.) TABLE 18.2 International Classification for Intraocular Retinoblastomas (Modified from National Cancer Institute: Retinoblastoma classification. Available at http://www.cancer.gov/cancertopics/pdq/treatment/retinoblastoma/HealthProfessional/page3.) A. Average age is 13 months, with 89% diagnosed before three years of age. A. Peters’ anomaly and retinoblastoma have occurred in the same neonate. XII. Human papillomavirus infection may be a cofactor in the development of retinoblastoma. II. “Two-hit” model B. In the hereditary form, the first mutation occurs in a germinal (prezygotic) cell (which, therefore, would mean that the mutation would be present in all resulting somatic cells), and the second mutation occurs in a somatic (postzygotic) neural retinal cell, resulting in multiple neural retinal tumors (multifocal in one eye, bilateral, or both), as well as in primary tumors elsewhere in the body (e.g., pineal tumors and sarcomas). A. If both chromosomal 13q14 regions are normal, no retinoblastoma will develop. E. Gain of chromosome 1q31–1q32 is found in >50% of retinoblastoma and is also common in other tumors. I. The retinoblastoma gene model may oversimplify the disease process. IV. Hereditary cases (approximately 40% of cases) D. A patient with sporadic disease has approximately a 12.5% chance of producing affected children. I. Strabismus and leukokoria are the most common clinical manifestations of retinoblastoma. III. Moderate lesions may present as: A. Leukokoria (i.e., “cat’s-eye reflex”; Figs. 18.1 and 18.2) B. “Pseudoinflammation”—that is, simulating uveitis, endophthalmitis, or panophthalmitis with or without pseudohypopyon (Fig. 18.3) 1. Any childhood intraocular inflammation should be considered retinoblastoma until proven otherwise. a. Diffuse vitreous seeding is associated with 16q24 loss. b. It also may be found in association with retinal astrocytoma. C. Iris neovascularization (rubeosis iridis; Figs. 18.5A and 18.5B) with a hyphema, chronic secondary closed-angle glaucoma, or both may be seen. Iris neovascularization can lead to hyphema. IV. Advanced lesions may present with proptosis (see Figs. 18.5C and 18.5D), distant metastases, or both. Although proptosis as a presenting sign is rare in the United States, it is relatively common in developing countries. Similarly, age at diagnosis and signs of more advanced disease, such as globe enlargement, correlate with the socioeconomic status of the patient’s country of residence. A. Multifocal growth (i.e., spontaneous development from more than one region of the same neural retina; Fig. 18.6) B. Bilateral involvement is itself a reflection of multifocal neural retinal involvement. C. Exophytic retinoblastoma (see Fig. 18.1) grows predominantly toward the subneural retinal space and detaches the neural retina. D. Endophytic retinoblastoma (see Fig. 18.2) grows predominantly toward the vitreous. The neural retina is not detached. E. Rarely, retinal neovascularization may be found with retinoblastoma. II. The basic cell type is the radiosensitive undifferentiated retinoblastoma cell (Fig. 18.7). A. Retinoblastoma cells seem to be neuron-committed cells that arise from photoreceptor progenitor cells or from primitive stem cells that are capable of differentiation along both neuronal and glial cell lines. 2. Others contend that retinoblastoma arises from mature neural cells and not from tumor stem cells. III. Rosettes are of two types: A. Flexner–Wintersteiner rosettes (Figs. 18.8 and 18.9; see also Fig. 18.7C) are the characteristic rosettes of retinoblastoma, but they are not always present. 1. In the rosettes, the cells line up around an apparently empty central lumen. 2. Special stains show that the lumen contains a hyaluronidase-resistant acid mucopolysaccharide. B. Homer Wright rosettes (see Fig. 18.8), named after John Homer Wright, are found in medulloblastomas, neuroblastomas, and, occasionally, retinoblastomas. IV. Pseudorosette—this very poor and confusing term is often used to refer to arrangements in the tumor that on cursory examination may resemble the aforementioned rosettes. The structures are formed by: B. Small foci of necrotic cells between larger masses of viable tumor cells C. Incompletely formed Flexner–Wintersteiner or Homer Wright rosettes V. Fleurettes and retinocytoma (Fig. 18.10; see also Figs. 18.8 and 18.9) B. Fleurettes may be absent, may be present in small nodules, or, rarely, may be present as the only cells in the tumor so that the entire tumor has differentiated into photoreceptor-like elements. 1. The fully differentiated retinoblastoma is called a retinocytoma (retinoma). 4. Multinucleated tumor cells may be found in retinocytomas. C. Those areas that show photoreceptor differentiation usually lack evidence of necrosis and show only occasional calcification. VI. Other histologic features include significant areas of necrosis (Fig. 18.11; see also Fig. 18.7), necrotic retinoblastoma presenting as orbital cellulitis, and necrosis leading to spontaneous and complete regression in a shrunken, scarred, calcific eye—a rare occurrence (Fig. 18.12), A. Calcification (see Fig. 18.11) is a frequent and important diagnostic feature. It is mainly present in areas of necrosis. Calcification begins in nonviable cells or cells that are undergoing necrosis. The calcification is intracellular and begins in the cytoplasm, probably in the mitochondria. B. Basophilic areas around blood vessels (Fig. 18.13), lying freely within the tumor, and on the lens capsule represent deposition of DNA liberated from necrotic retinoblastoma cells. VII. Mode of extension A. Local spread 1. Anteriorly by seeding, into the vitreous and aqueous (see Figs. 18.2B and 18.3) 2. Posteriorly (see Figs. 18.5C and 18.5D) by direct extension into the subneural retinal space (Fig. 18.14) 1. Orbit (see Figs. 18.5C and 18.5D) 2. Brain C. Metastases (Fig. 18.15; see section Prognosis, later) The retinoblastoma five-year survival rate in the United States from 1975 to 1984 was 92.3%; it increased to 93.9% for 1985–1994 and to 96.5% for 1995–2004. I. Metastases—factors that appear to be independently associated with the development of metastases include: A. Invasion of the cut end of the optic nerve—see later B. Invasion of the choroid—see later C. Enucleation of the globe more than 120 days after initial diagnosis: five-year metastatic risk, 4% II. Bilateral and unilateral cases have the same fatality rate. B. When choroidal invasion is slight (most cases with choroidal invasion), the mortality rate appears not to be affected; when the invasion is massive (Fig. 18.16), the mortality rate is approximately 60%. C. Optic nerve involvement (see Fig. 18.16B) 1. When the optic nerve is not invaded, the mortality rate is approximately 8%. D. The presence of iris neovascularization (rubeosis iridis) is a poor prognostic sign. E. Probably only retinoblastoma patients with the genetic (familial) tumor or sporadic tumors with germinal cell gene mutations have a definite predilection for the development of fatal second malignancies (with or without radiation therapy for the initial lesion), including a pineal gland tumor, despite adequate control of their original eye tumor.
Retinoblastoma and Pseudoglioma
Retinoblastoma
General Information
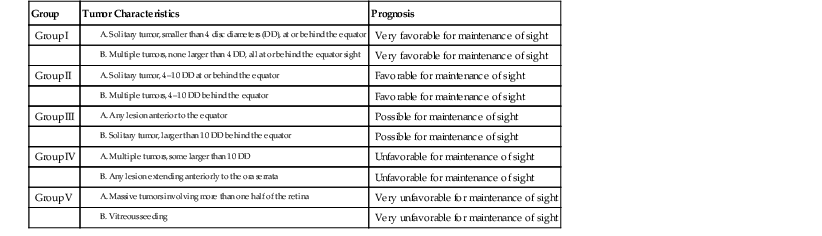
Group
Tumor Characteristics
Prognosis
Group I
Very favorable for maintenance of sight
Very favorable for maintenance of sight
Group II
Favorable for maintenance of sight
Favorable for maintenance of sight
Group III
Possible for maintenance of sight
Possible for maintenance of sight
Group IV
Unfavorable for maintenance of sight
Unfavorable for maintenance of sight
Group V
Very unfavorable for maintenance of sight
Very unfavorable for maintenance of sight

Heredity
Knudson’s two-hit model states that retinoblastoma arises as a result of two mutational events involving the RB1 gene (see discussion of chromosomal region 13q14, later).
Clinical Features
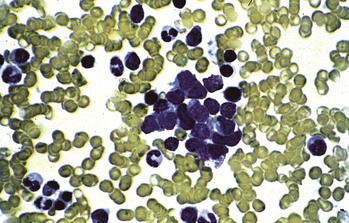
Histology
In Homer Wright rosettes, the cells line up around an area containing cobweb-like material but no acid mucopolysaccharide.
The most common site of metastasis is the central nervous system, and it has an extremely poor prognosis, particularly if radiotherapy is not utilized in the treatment.
Prognosis
Overview
Choroidal invasion is a risk for metastases, especially if the invasion is associated with iris neovascularization, increased intraocular pressure, or optic nerve invasion.
Retinoblastoma and Pseudoglioma
18
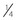 of the retina
of the retina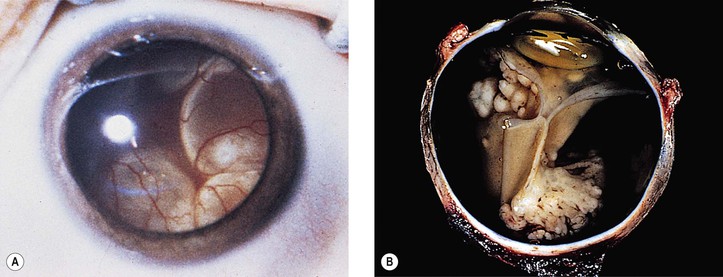
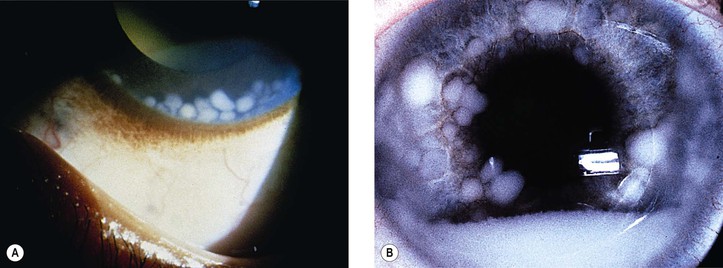
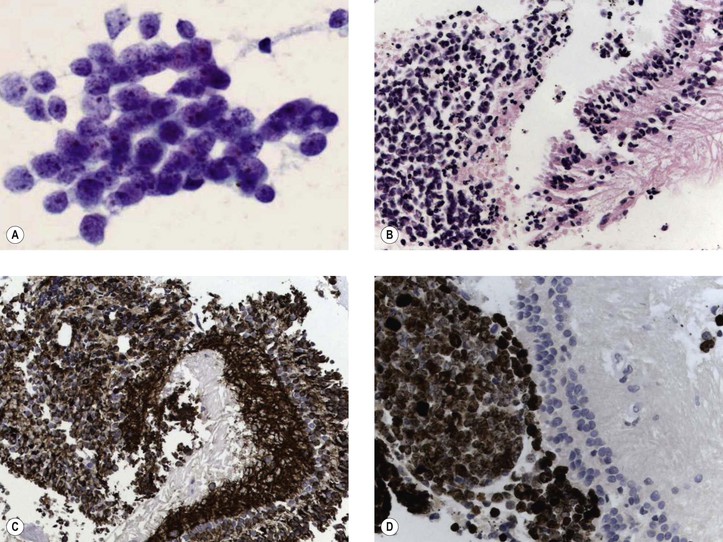
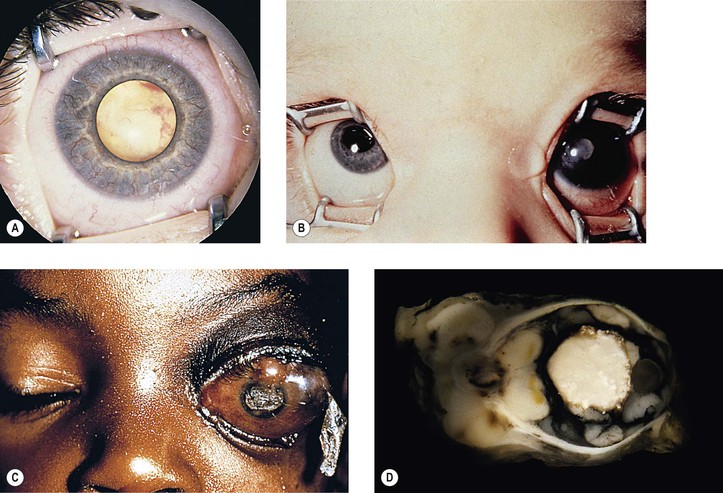
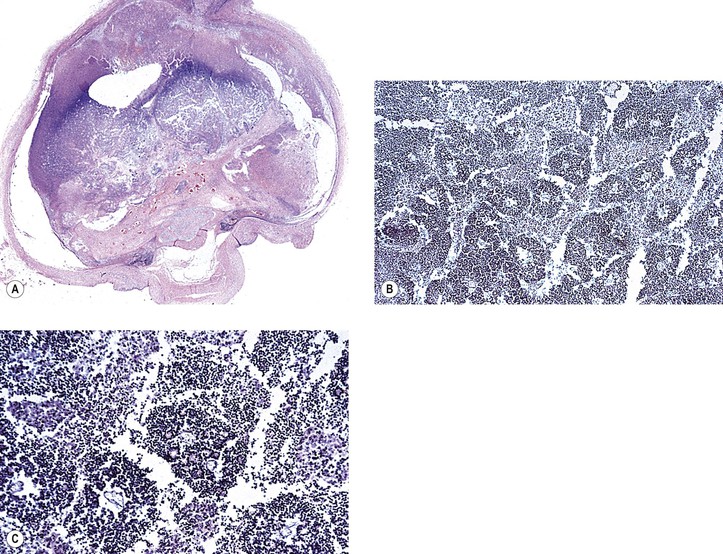
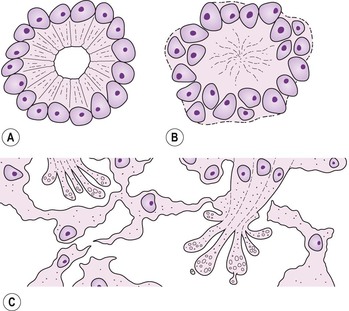
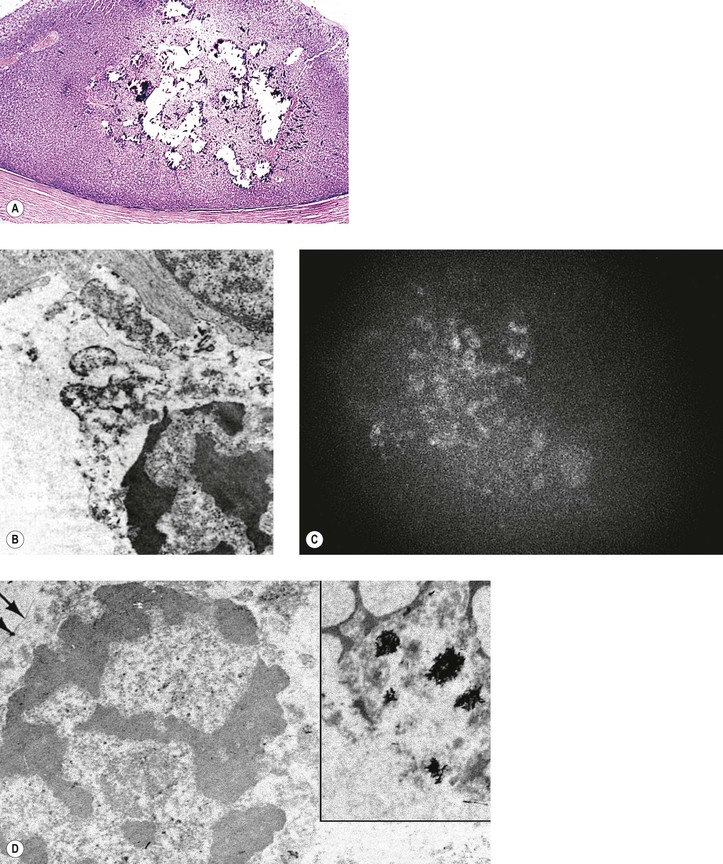
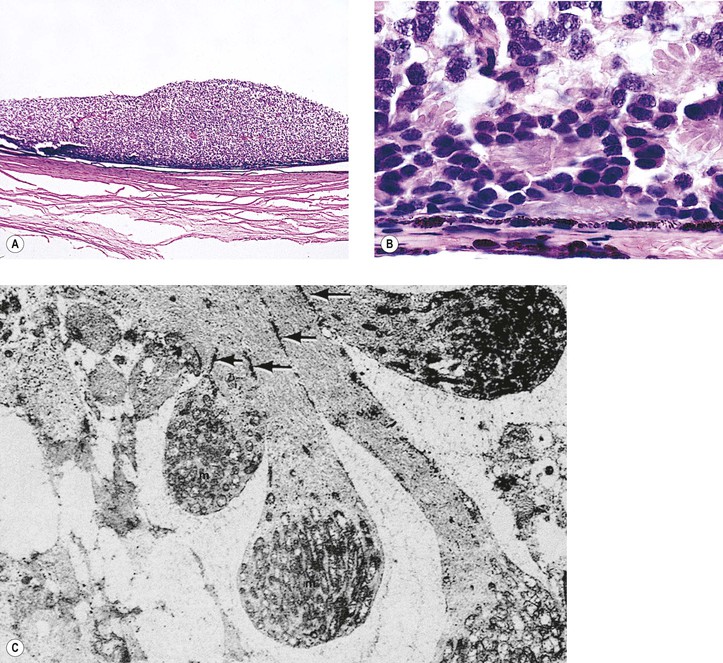
Fig. 18.10 Fleurettes. A, Retinoblastoma shows complete photoreceptor differentiation (see Fig. 18.6 for fundus and gross appearance). B, Note arrangement (fleurettes) of cells that have undergone photoreceptor differentiation. C, Photoreceptor inner segments resemble those of cone cells. They radiate from attachment girdle (zonulae adherentes) (arrows) of external limiting membrane, partly because intervening Müller cell processes are lacking (m, mitochondria). (C, Modified from Tso MOM, Zimmerman LE, Fine BS: The nature of retinoblastoma: I. Photoreceptor differentiation. Am J Ophthalmol 69:339. © Elsevier 1970.)