18 RETINA AND VITREAL DISEASE
Retinal Tear and Rhegmatogenous Retinal Detachment
W. Craig Lannin
ICD-9: 361.32—HORSESHOE TEAR OF RETINA WITHOUT DETACHMENT
ICD-9: 361.0—RETINAL DETACHMENT WITH RETINAL DEFECT
 THE DISEASE
THE DISEASE
Significant ocular morbidity is associated with retinal detachment (RD) and to a lesser extent retinal tear. As typical symptoms usually herald the possible development of a retinal tear, educating at-risk patients about the nature and significance of such symptoms is an important means of reducing the incidence of progression to rhegmatogenous detachment. Compared to RD surgery, the treatment of retinal tears is less expensive, requires less rehabilitation, is technically less demanding, results in fewer serious complications, and is much less likely to be associated with significant visual loss.
Pathophysiology
RD can result from a variety of mechanisms. Rhegmatogenous RDs result from the passage of fluid from the vitreous cavity through a hole or tear in the retina into the subretinal space. Although round atrophic holes may lead to RD, the majority of clinically significant RDs result from retinal tears related to vitreoretinal traction. An important event in the genesis of such tears is posterior vitreous detachment (PVD).
Retinal tears often occur along the posterior border of the vitreous base or at other sites of vitreoretinal adhesion, following spontaneous or traumatic PVD. After the vitreous has separated from its broad contact with the retina, which usually follows in short order after the attachment at the optic disc is broken, vitreoretinal traction is increased at these sites of adhesion. Vitreous movement initiated by rapid head turning or eye rotation is then translated through force vectors to these focal areas of residual vitreoretinal attachment and may lead to the formation of a retinal tear.
Greater than average vitreoretinal adherence may be associated with developmental anomalies, including cystic retinal tufts and localized posterior extensions of the vitreous base. Exaggerated adherence is also often found along the course of retinal blood vessels, which is reflected in the frequent association of vitreous hemorrhage with acute retinal tear. Finally, firm vitreoretinal adhesions are associated with obvious areas of retinal alteration, including retinal lattice degeneration and sites of previous inflammation.
In order for a rhegmatogenous RD to develop, there must be a break in the retina and relatively fluid vitreous overlying the break. PVD increases the probability of occurrence for both of these conditions. Initially, the subretinal fluid is composed of fluid originating in the vitreous cavity, but this is most likely augmented by serum derived from the choriocapillaris as time passes. Variables that influence whether fluid does gain access to the subretinal space include the amount of traction being exerted on the edges of the break, the effects of gravity (location of the break above or below the horizontal midline), and the lifestyle and activities of the affected person.
Etiology
Risk factors for retinal tear and detachment include myopia, a history of intraocular surgery, ocular trauma, lattice degeneration, a familial history of tear/detachment, and a history of either condition having occurred in the fellow eye. About half of all patients who have an RD are myopic. Historically, cataract surgery was known to increase the risk of RD. This risk has been mitigated to a significant extent by small incision surgery and other technical improvements. A history of posterior capsular rupture and vitreous loss increases the risk of subsequent detachment. YAG laser capsulotomy has also been identified as an independent risk factor.
The Patient
Clinical Symptoms
- Symptoms of an acute retinal tear include a sudden onset of floaters and debris, photopsia, and blurred vision. These same symptoms can occur in uncomplicated PVD. Photopsia associated with vitreoretinal traction is typically of very short duration, unlike the scintillations encountered with migraine or other vascular disturbances.
- When an RD has occurred, patients may additionally become aware of visual field loss, with the perception of a mobile shadow or curtain obscuring their vision. If the macula has become involved, there will usually be significant loss of central visual acuity as well. Patients with chronic, slowly progressive, or demarcated detachments, usually in the inferior retina, may be asymptomatic.
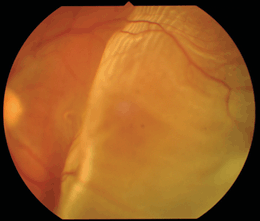
Figure 18-1. Retinal detachment. (Photo courtesy of Leonid Skorin Jr.)
Clinical Signs
See Figure 18-1.
- As noted above, PVD is usually a precursor of retinal tear and detachment and is evident clinically in most cases. Slit-lamp exam may reveal the presence of fine pigment granules in the anterior vitreous, which, in the absence of a history of prior intraocular surgery, is highly suggestive of a retinal break. When a vitreous hemorrhage occurs in association with PVD, a retinal tear will be present about 70% of the time.
- Binocular indirect ophthalmoscopy with scleral depression or peripheral contact lens examination will usually reveal the attached flap of a retinal horseshoe tear under persistent vitreoretinal traction, or a free-floating operculum overlying a retinal hole if the traction has been alleviated. The tear itself usually has a darker orange color than the surrounding retina due to the unimpeded illumination of the underlying choroid.
- The most obvious visible evidence of the presence of a detached retina is its elevation from the surface. The surface of the detached retina has a corrugated and often dimpled appearance and demonstrates movement with motion of the head and/or eye. This mobility may be significantly reduced in long-standing RDs or in those complicated by proliferative vitreoretinopathy (PVR).
- The configuration of the detachment is an important clue to help pinpoint the location of the retinal break(s). With a superior detachment, the retinal break is usually at or near the 12 o’clock position. When a detachment of one inferior quadrant has occurred, a break will almost always be found superior to a line radially bisecting the area of detachment. In an inferior detachment, if the subretinal fluid extends higher on one side than the other, the break will be found on the higher side. About half of all RDs are associated with a single break, but the possibility of multiple breaks must always be kept in mind.
- Confrontation testing will often demonstrate a visual field defect, and Snellen acuity will be reduced if the macula is detached. Tonometry will usually reveal a relatively low intraocular pressure (IOP) in an eye with a RD, although rarely the pressure may actually be higher than normal. Pupillary testing will often demonstrate a relative afferent pupillary defect (RAPD), although this is typically much less prominent than that encountered with optic nerve disease or with central retinal vascular occlusion. Ultrasonography and occasionally electroretinography (ERG) may be helpful in cases where media opacity prevents adequate visualization of the retina.
- The configuration of the detachment is an important clue to help pinpoint the location of the retinal break(s). With a superior detachment, the retinal break is usually at or near the 12 o’clock position. When a detachment of one inferior quadrant has occurred, a break will almost always be found superior to a line radially bisecting the area of detachment. In an inferior detachment, if the subretinal fluid extends higher on one side than the other, the break will be found on the higher side. About half of all RDs are associated with a single break, but the possibility of multiple breaks must always be kept in mind.
Demographics
Approximately one of every 15,000 individuals has an RD in any given year. The incidence of retinal break is significantly greater, about 6%, but the majority of these breaks are small atrophic holes with a low potential for progression to RD. Historically, the peak incidence of RD has occurred between 50 and 70 years of age, with the frequency in that age group largely reflecting the incidence of both PVD and, to a lesser extent, cataract surgery. The current trend of fewer cataract surgery–related detachments may alter these numbers. Myopic detachments are most common between ages 25 and 45. There is another smaller peak in the teenage years, and these detachments are frequently of traumatic origin.
About 60% of RD patients are males, and trauma does not completely account for this propensity. Although familial patterns of RD are sometimes evident, most are sporadic. A large part of the familial risk profile is of an indirect nature, such as a genetic predisposition to myopia and/or lattice degeneration.
Horseshoe retinal tears occur with the greatest frequency in the superotemporal retina (~60%), followed by the superonasal quadrant. Traumatic retinal dialyses have a propensity to occur in the inferotemporal retina, particularly in the young. Retinal breaks occur at or anterior to the equator in about 85% of cases. Of all rhegmatogenous detachments, 50% have more than one break. The risk of RD in the second eye of an affected individual has been estimated to be about 10% to 15% in phakic individuals and 25% to 40% in aphakic or pseudophakic individuals.
Differential Diagnosis
The differential diagnosis of rhegmatogenous RD includes exudative RD, traction RD, retinoschisis, retinal/choroidal tumor, and choroidal detachment. A thorough history and careful examination usually make the distinguishing characteristics of these entities evident. Exudative detachment is characterized by shifting subretinal fluid, and the underlying inflammatory or other causative disorder is usually also apparent. Traction detachment is associated with a concave retinal configuration and lack of significant movement. Choroidal tumors and detachment can be distinguished from RD by their smooth contour and the presence of retinal pigment epithelium (RPE) and choroidal markings associated with the elevated retina.
The Treatment
Retinal Tear
- Treatment for an acute, symptomatic flap tear is justified as a means of reducing the risk of progression to RD. Studies have shown such tears to progress to detachment in approximately one third of cases. Treatment for symptomatic operculated holes is also justifiable, although the danger of progression is less than for a flap tear with persistent vitreoretinal traction.
- In asymptomatic patients, the decision whether or not to treat must be individualized, taking into account multiple factors. These include the location, type, and size of the break(s), with superior breaks, flap tears, and large breaks being considered for prophylaxis. If there is pigmentation around the break, it is not acute and treatment may not be indicated. If there is significant localized subretinal fluid around the break, treatment may be indicated. The presence of vitreous hemorrhage increases the risk associated with a retinal break.
- Other factors that must be weighed in the decision are the age and life expectancy of the patient, the patient’s lifestyle, the status of the fellow eye, the refractive error (in particular, the degree of axial myopia), the presence of aphakia or pseudophakia, anticipated cataract surgery, and the family history. The timing of treatment is dictated by the apparent danger associated with a particular break.
- Prophylactic treatment of retinal breaks is usually accomplished using either transconjunctival cryopexy or laser photocoagulation. The choice of technique is guided by the number, size, and position of the break(s), as well as by the preference of the patient and surgeon.
- There is some theoretical advantage of laser over cryotherapy due to the possibility of dispersing viable RPE cells with cryotherapy. It is felt by many surgeons that there is a greater risk for the subsequent development of an epiretinal membrane (ERM) and associated macular pucker associated with cryotherapy. There has thus been a trend toward increased utilization of laser for prophylactic treatment. However, there are some cases where cryotherapy still holds a distinct advantage, such as cases of very anterior breaks and when visualization is impaired due to the presence of significant cataract or vitreous hemorrhage.
- Other than in cases of giant tear, scleral buckling (SB) is rarely utilized for prophylactic treatment.
- Laser photocoagulation of retinal tears can usually be accomplished with only topical anesthesia, unless extensive treatment of multiple breaks or lattice degeneration is contemplated. Transconjunctival cryopexy is generally performed utilizing topical anesthesia augmented with a subconjunctival injection in the area overlying the break(s).
- Whichever modality is chosen, the goals of treatment are the same: to insure an adequate width of treatment that completely surrounds the retinal break. The resultant scar will then isolate the break and prevent fluid from the vitreous cavity from gaining access to the subretinal space.
- Following surgery, the patient is instructed to restrict activity until an effective adhesion has developed, usually 7 to 10 days for cryopexy or a slightly shorter period of time for laser.
- Prophylactic treatment of high-risk retinal breaks has been shown to be effective in significantly reducing the risk of subsequent RD. Serious complications are rare, with the most frequent visually significant adverse consequence being macular pucker. However, macular pucker may also develop in association with retinal breaks that have not been treated.
Retinal Detachment
- The decision to treat an RD must also be individualized, taking into account the likelihood of progression, the residual visual potential, the age, lifestyle, and life expectancy of the patient, the status of the fellow eye, and the patient’s wishes.
- When an acute, macula-threatening, or recent macula-off detachment is encountered, surgery is indicated as soon as possible. Appropriate bedrest and positioning instructions are given to reduce the possibility of extension of the detachment into the macula, if it is still attached. This may also allow for significant absorption of subretinal fluid after RD surgery.
- An inferior demarcated detachment may not require surgery. Likewise, the limited visual prognosis associated with a long-standing macula-off detachment may persuade the patient and surgeon not to pursue surgical intervention, particularly if there is excellent vision in the contralateral eye.
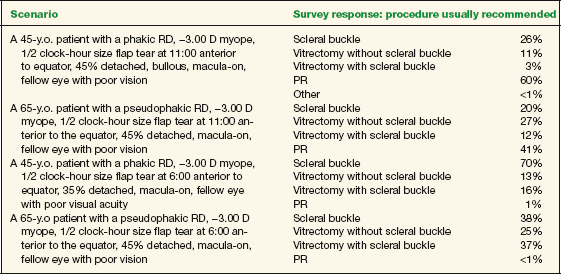
- Available methods of retinal reattachment surgery include SB, pars plana vitrectomy (PPV), pneumatic retinopexy (PR), and a temporary balloon buckle. The American Society of Retina Specialists (ASRS) polled its membership about preferences for method of surgical repair of RD in the 2006 PAT (Preferences and Trends [PAT]) survey. Several typical scenarios were described, with differing patient characteristics that often influence the choice of surgical technique. The four scenarios and survey results are shown in Table 18-1.
- Traditionally, the surgical technique most commonly employed in the treatment of rhegmatogenous RD has been SB, in combination with retinopexy utilizing cryotherapy or laser photocoagulation.
- The purpose of the buckle is to aid in bringing the treated breaks back into contact with the RPE, and to relieve persistent vitreoretinal traction. The scleral buckle is created utilizing either solid silicone rubber elements or soft silicone sponges, which are permanently anchored to the sclera with sutures.
- Placement of the buckle can be segmental, with either radial or circumferential elements, or encircling, passing under the four rectus muscles. In some cases, a combination of an encircling band or tire is combined with a radial sponge.
- Cryotherapy or laser is necessary in order to produce a controlled inflammatory reaction that will result in a permanent adhesion of the retina once it is brought back into contact with the RPE. Laser cannot be effectively employed until after the retina has been brought back into close proximity with the pigment epithelium, whereas cryotherapy can be applied around the retinal breaks in the presence of subretinal fluid.
- Drainage of subretinal fluid is often required in order to bring the retina into contact with the buckle and also to provide space to achieve an adequate buckle height without an excessive elevation of IOP. The need for drainage is influenced by several factors, including the amount of retinal elevation, the number, size, and position of retinal breaks, and the duration of the detachment, as long-standing subretinal fluid becomes highly viscous and resistant to reabsorption.
- A successful outcome of surgery is dependent on finding and closing all the retinal breaks, not necessarily only the causative break.
- Surgery is usually performed under regional block anesthesia, although general anesthesia may be necessary in young patients or if a lengthy procedure is a possibility. Some surgeons prefer general anesthesia for most cases.
- PPV has become the method of choice for treating RDs associated with giant retinal tears and for dealing with other complicated scenarios. Some surgeons now prefer PPV as their first option in the primary repair of most RDs. Vitrectomy is also necessary to repair recurrent RDs associated with significant PVR.
- The vitrectomy is often supplemented with adjunctive techniques, including the use of membrane dissection, internal drainage of subretinal fluid, laser, fluid-gas exchange, intraocular silicone oil, and, particularly in the treatment of giant tears, liquid perfluorocarbon. In many cases, it is also necessary to employ a scleral buckle.
- PPV has become increasingly popular, particularly in Europe, as a primary procedure for rhegmatogenous RD. In the United States, many surgeons employ primary PPV for pseudophakic detachments, where its tendency for cataractogenesis is a moot point. There has been a significant recent trend in vitreous surgery to employ smaller gauge instrumentation, with 23-gauge and 25-gauge systems increasingly utilized.
- Reported advantages of PPV over SB include better intraoperative control, the avoidance of a very soft eye during surgery, the avoidance of drainage complications, and a lower incidence of refractive change and diplopia. Disadvantages include the invasive nature of the surgery and the associated risk of endophthalmitis, cataractogenesis in phakic patients, the higher rate of iatrogenic and missed/new breaks, more frequent persistent postoperative IOP elevation, the more technologically intense nature of the procedure requiring more skilled assistance and nursing, and probably its increased expense.
- PR is an effective method of repair, if cases are selected carefully. It is more successful when limited to detachments with superior breaks and to cases with a single break or breaks which are minimally separated.
- The technique consists of a pars plana injection of perfluorocarbon gas, again combined with cryopexy and/or laser photocoagulation. The patient must be willing and able to sustain a strict postoperative positioning regimen, or the chances of success are dramatically reduced. The head is positioned so as to bring the position of the break to the 12:00, so that the gas bubble completely covers the break. This must be continued until the break has been brought back into contact with the RPE, and a good cryopexy or laser scar is developing.
- In general, better results have been obtained in phakic patients. PR is contraindicated in the presence of extensive lattice degeneration or significant PVR.
- Gas injection may also be utilized in conjunction with SB, in order to facilitate the reabsorption of subretinal fluid.
- The placement of a temporary balloon buckle is also an effective method of repairing selected RDs, although its use never became widespread in the United States. Similar to PR case selection, the temporary balloon buckle is almost always limited to detachments with a single break. Unlike PR, the balloon technique can be employed to treat an inferior break. Currently, the material is not commercially available.
- Approximately 80% to 85% of primary RDs can be successfully repaired utilizing a single SB operation, and approximately 92% of detachments overall can be successfully repaired, including those that require two or more procedures.
- The success rates for PPV are similar to those of SB in uncomplicated cases, and possibly superior in some hands. The success rates for PPV in complicated cases of RD are significantly higher than with SB.
- The one-operation success rates with PR are somewhat less than with either SB or PPV, but failed attempts at this procedure do not seem to significantly jeopardize the ultimate final outcome. An important advantage of PR is its significantly lower overall costs relative to SB and PPV, which must be performed in the operating room. There may also be faster rehabilitation with PR, at least in some cases. When careful case selection is adhered to, the use of this modality can be advocated.
- Visual results following RD surgery are in large part determined by the preoperative visual acuity and the status of the macula. Overall, 55% of anatomic surgical successes will achieve at least 20/50 vision within 6 months of surgery, but about 15% will be left with 20/400 or worse. The average long-term visual result for a repaired macula-off detachment is about 20/70 acuity. A patient with a macula-on detachment should be advised preoperatively that the cost of repairing the detachment may be a mild decrease in visual acuity, even with successful surgery.
- Patients with a retinal tear and/or a RD require careful follow-up to watch for the development of complications and to monitor the status of the fellow eye.
- The most common cause of failed surgery is the development of PVR. PVR is characterized by the development of fibrous cellular proliferation that leads to the formation of epiretinal and/or subretinal membranes that distort the retinal surface into fixed folds and often reopen existing breaks or cause new ones to form. Its development portends a relatively poor prognosis, particularly in regards to the visual outcome.
- Other significant complications include intraoperative subretinal hemorrhage, retinal incarceration, and new retinal break formation, and postoperative choroidal detachment, diplopia, strabismus, cystoid macular edema (CME), and macular pucker.
- The purpose of the buckle is to aid in bringing the treated breaks back into contact with the RPE, and to relieve persistent vitreoretinal traction. The scleral buckle is created utilizing either solid silicone rubber elements or soft silicone sponges, which are permanently anchored to the sclera with sutures.
TABLE 18-1 Selected Results from the 2006 ASRS PAT Survey
Degenerative Retinoschisis
W. Craig Lannin
ICD-9: 361.10
 THE DISEASE
THE DISEASE
Although retinoschisis occurs in both hereditary (X-linked), juvenile and acquired, degenerative varieties, this discussion is limited to the much more common degenerative form. Degenerative (age-related, senile) retinoschisis is a relatively rare precursor to clinically significant (progressive) retinal detachment (RD), but can sometimes be difficult to distinguish from a chronic detachment. This differentiation is one of the most clinically important aspects of retinoschisis.
Pathophysiology
Degenerative retinoschisis represents a splitting of the retina, usually originating in the outer plexiform layer, and develops as a progression of peripheral cystoid degeneration.
Cystoid degeneration is found in the vast majority of adult eyes and begins with the development of tiny spaces within the outer plexiform layer. As the cysts enlarge, the spaces are bridged by glial cell fibers that extend from the external to the internal limiting membranes (ILMs). This process begins near the ora serrata and extends circumferentially and posteriorly but usually not posterior to the equator. Based on pathologic and, less distinctly, clinical features, cystoid degeneration may be subdivided into typical and reticular forms. The much less common reticular form has a greater potential to spread more posteriorly, beyond the equator.
With further enlargement of the cysts, the stretched glial pillars may eventually rupture, resulting in the development of retinoschisis. The inner wall of the schisis cavity typically consists of the ILM, the nerve fiber layer (NFL), the retinal vessels, and the inner plexiform layer. The outer wall is composed of degenerated elements of the inner nuclear, outer plexiform, and outer nuclear layers, with a relatively well-preserved photoreceptor layer. The cavity itself contains a viscous, hyaluronidase-sensitive mucopolysaccharide substance. The margins of the schisis are contiguous with areas of cystoid degeneration.
Degenerative retinoschisis may also be divided into typical and reticular forms. Both types are usually found in the temporal retina, particularly the inferotemporal quadrant, although the reticular form is also seen in the nasal retina less rarely than is the typical form. Reticular retinoschisis is more often highly bullous and frequently has an extremely thin inner wall. Reticular schisis also usually extends posterior to the equator, unlike typical schisis, and may rarely even threaten the macula. As stated above, cystoid degeneration is almost universal in adult eyes, but retinoschisis is much less common. It is not certain why in some cases cystoid degeneration progresses to degenerative retinoschisis. There does not appear to be a genetic predisposition to this process. Bullous retinoschisis seems to be more common when zones of typical and reticular cystoid degeneration overlap.
Some authors hold that the precipitating factor is an age-related degenerative change in the inner retinal layers related to vascular insufficiency and impaired cellular nutrition. This theory is supported by the increasing prevalence of retinoschisis with advancing age and by the association of vascular abnormalities seen with some cases of the condition. Other authors believe that vitreoretinal traction plays a role in the development and progression of retinoschisis. However, studies have failed to demonstrate that vitreoretinal relationships play a significant role in these events, and posterior vitreous detachment (PVD) in the presence of retinoschisis only rarely results in the development of a progressive, symptomatic RD.
The Patient
Clinical Symptoms
- The vast majority of patients (~99%) with retinoschisis are asymptomatic. Although retinoschisis causes an absolute visual field defect, most patients are unaware of these, except in those rare cases where the schisis extends posteriorly into the macular region. The reasons for the usual lack of recognition of the field cut include the typically very slow or lack of progression of the retinoschisis and the fact that nasal field loss associated with the usual temporally located schisis is often overlooked.
- Complications of retinoschisis include RD and vitreous hemorrhage. The localized, nonprogressive type of RD does not usually cause symptoms, but progressive, clinically significant detachments obviously do. Vitreous hemorrhage, causing an acute onset of floaters, or hemorrhage into the schisis cavity may rarely be associated with retinoschisis, but such hemorrhages are usually minor. Incidental PVD is a more common source of both vitreous hemorrhage and floaters in general.
Clinical Signs
See Figure 18-2.
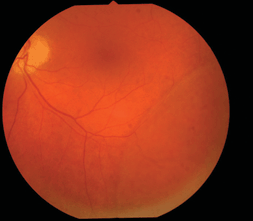
Figure 18-2. Degenerative retinoschisis. (Photo courtesy of Leonid Skorin Jr.)
- The inner layer of the retinoschisis is visible as a smooth-domed elevation containing the retinal vessels. The retinal vessels may appear white due to sheathing. Retinoschisis is found predominantly in the temporal retina and is usually bilateral, although it may be asymmetric in extent and elevation. The schisis cavity may increase or decrease in height over time, and complete disappearance has been noted in approximately 2% of cases. Tiny glistening flecks are often seen on the inner layer, although these have little clinical significance. New areas of schisis may develop elsewhere in about 10% of cases. Posterior extension of the retinoschisis may occur, although involvement of the macula is rare. Most cases stop within 3 disc diameters of the macula.
- Holes may develop in either or both of the walls of a retinoschisis cavity in about 15% of cases, with outer layer holes much more common than inner layer holes. Clinically, what appears to be an inner layer hole may actually represent only an area of extreme thinning. When an inner layer hole does develop, it is usually quite small and difficult to appreciate clinically. Outer layer holes are usually much more obvious, as they tend to be large. They have a rounded contour, may be single or multiple, and are seen typically in association with reticular retinoschisis. Outer layer holes often have a posteriorly rolled edge. Fundus contact lens examination is useful in the recognition of outer and inner layer holes.
- Although progressive rhegmatogenous RD is the most important clinical complication of degenerative retinoschisis, this is actually rare, accounting for probably no more than 2.5% of all detachments. In about 6% of cases of retinoschisis, localized, asymptomatic, nonprogressive, or very slowly progressive RDs develop, and these do not extend much beyond the border of the area of schisis. There may be a fine demarcation line present if one of these localized subclinical RDs is present. All schisis-related RDs are associated with outer layer holes. The viscosity of the fluid within the retinoschisis cavity is probably an important factor in inhibiting the progression of schisis-related detachments with outer layer holes. Inner layer holes or traction-related tears are also sometimes present, particularly with progressive, symptomatic RDs.
- The differential diagnosis of degenerative retinoschisis includes RD, choroidal detachment/tumors, and juvenile (X-linked) retinoschisis. The latter two groups are usually easily excluded with a thorough history and careful examination. In some cases, the differentiation of retinoschisis from a chronic RD can be challenging. Table 18-2 illustrates some distinguishing characteristics, which are helpful in this distinction.
- Although progressive rhegmatogenous RD is the most important clinical complication of degenerative retinoschisis, this is actually rare, accounting for probably no more than 2.5% of all detachments. In about 6% of cases of retinoschisis, localized, asymptomatic, nonprogressive, or very slowly progressive RDs develop, and these do not extend much beyond the border of the area of schisis. There may be a fine demarcation line present if one of these localized subclinical RDs is present. All schisis-related RDs are associated with outer layer holes. The viscosity of the fluid within the retinoschisis cavity is probably an important factor in inhibiting the progression of schisis-related detachments with outer layer holes. Inner layer holes or traction-related tears are also sometimes present, particularly with progressive, symptomatic RDs.
TABLE 18-2 Clinical Signs: Retinoschisis Versus Retinal Detachment

Demographics
The prevalence of retinoschisis is 3% to 4% in patients over age 10 and 7% in patients over 40. No significant differences in the incidence of degenerative retinoschisis based on racial, sexual, or geographic factors have been demonstrated. Degenerative retinoschisis is more common in hyperopic eyes. Retinoschisis is bilateral in more than 80% of cases. The risk of progression to clinical RD is approximately 1 in 2000.
Treatment is rarely required for degenerative retinoschisis. Only in those rare cases when progressive, clinically significant RD develops is surgery indicated. This usually entails cryotherapy and scleral buckle (SB) to close the outer wall breaks, most often with drainage of subretinal fluid. However, if the outer layer breaks are posterior, multiple, and/or large, cryotherapy with drainage and intraocular gas injection or vitrectomy techniques may be required. Patients with localized detachments, particularly those with demarcation lines, may be managed conservatively with periodic follow-up and education about the warning symptoms of progressive RD.
Prophylactic treatment of outer layer holes is of dubious value, as progressive RD rarely occurs and treatment is not without risk. This may be justified in cases where there has been a schisis-related detachment in the opposite eye.
Treatment of posterior extension of retinoschisis is rarely indicated, as the risk of macular involvement is quite small, and again treatment may be associated with complications.
All patients with retinoschisis should be followed-up at least every 6 to 12 months, depending on the duration and stability of the condition. They should be instructed to call promptly in the event of the development of symptoms suggestive of retinal tear or detachment.
Posterior Vitreous Detachment
Michael J. Trad
ICD-9: 379.21
 THE DISEASE
THE DISEASE
Pathophysiology
Posterior vitreous detachment (PVD) represents a cortical vitreous and hyaloid membrane detachment from the posterior retina. PVD occurs as a result of anterior contractional forces exerted by the overlying fibrous vitreous at the vitreous base, secondary to fibrillary degeneration and hyaloid membrane thinning. Fibrillary degeneration encompasses both syneresis (vitreal shrinkage) and liquefaction of vitreous. Gravitational forces also play a role in the inferior and anterior collapse of the vitreal body. However, PVD may also occur without collapse, although this is less common. PVD also is termed complete when it extends to the ora serrata or incomplete when involving only specific regional areas.
PVD is of greatest clinical concern as a result of its potential to cause retinal tears at preexisting sites of increased vitreoretinal adherence (lattice degeneration, retinal tuft, etc.). This tractional influence may result in retinal hemorrhage and tear; vitreous hemorrhage (those associated with PVD are usually small and self-limiting); vitreomacular traction syndrome; macular edema, hole, or epiretinal membrane (ERM); and retinal detachment (RD) (secondary to continuous flow of liquid vitreous through retinal breaks).
Etiology
PVD typically occurs as a normal consequence of aging but can also be associated or potentially linked with trauma, chorioretinitis, myopia, vigorous exercise or isolated head movement, heavy lifting, aphakia, and pseudophakia.
The Patient
Clinical Symptoms
- Floaters
- Transient photopsia
- Blurred vision
- Metamorphopsia
Clinical Signs
Prominent Signs
- Prepapillary floater in the shape of an annular ring, line, or other geometric form
- Occasionally, preretinal and vitreous hemorrhage, retinal hemorrhage, retinal tear, and RD subtle signs
- Macular edema
- Macular hole
- Pseudo-operculum anterior to macula
- ERM
- Retinal striations
- White without pressure
- Schafer’s sign (tobacco dust)–pigmented particles (red blood cells or retinal pigment) in anterior vitreous
- Peripheral retinal operculum (if associated with a retinal break)
- Macular edema
Demographics
- PVD rarely occurs in patients younger than 45 years of age unless associated with chorioretinitis or trauma.
- Vitreous detachment of the optic disc is present in approximately one-half of all patients older than 50 years of age.
- By age 65, approximately 31% of the population has experienced a complete PVD.
- Complete PVD is much more common than incomplete PVD.
- Up to 50% of PVDs are associated with photopsia.
- Approximately 90% of patients who report concurrent flashes and floaters have PVD, and 11% have retinal breaks.
- Retinal breaks occur in up to 15% of patients with symptomatic PVD.
- 7.5% of PVDs have an associated vitreous hemorrhage.
- Traction at the posterior pole resulting in macular edema occurs in about 2.5% of symptomatic PVDs.
Significant History
- Trauma
- Chorioretinitis
- May potentially be precipitated by aphakia, pseudophakia, myopia, heavy lifting, and vigorous exercise or isolated head movement
Ancillary Tests
Ocular assessment for PVD may include confrontation visual fields, Amsler grid, biomicroscopy, and dilated fundus evaluation.
The Treatment
No treatment for isolated PVD is necessary. Occasionally, Nd:YAG laser vitreolysis or pars plana vitrectomy (PPV) is employed for patients intolerant of floaters. Patients with no apparent retinal involvement at the time of initial presentation must be thoroughly educated as to the necessity to return immediately upon recognition of new floaters or flashes. It is this early stage in which symptomatology is detectable that is vital in providing a therapeutic window of opportunity for the prevention of RD. Differing opinions prevail as to both the usefulness and frequency of regular follow-up visits because retinal tears may occur anywhere from a few weeks to a decade after initial examination of uncomplicated PVD.
Idiopathic Epiretinal Membranes
Michael J. Trad
ICD-9: 362.56
 THE DISEASE
THE DISEASE
Pathophysiology
Idiopathic epiretinal membranes (IERMs) are nonvascular proliferative lesions of the macular or paramacular vitreoretinal interface and typically develop unilaterally in individuals ≥50 years of age. IERMs are believed to develop secondary- to venous-flow impedance at retinal arteriosclerotic arteriovenous (AV) crossing, posterior vitreous detachment (PVD), and/or subclinical focal retinal inflammation. These conditions may disrupt the retina’s internal limiting membrane (ILM) at sites where it is thin and firmly adherent (blood vessels and optic disc), stimulating a reparative process in which Muller cell processes and astroglial cells migrate through ILM breaks to proliferate on the retinal surface (within the subhyaloid space). Other IERMs that are composed of collagen (“cortical vitreous preretinal membranes”) or a combination of collagen and retinal glial cells may also occur.
IERMs typically display little or no progression. However, some may grow and contract enough to cause irregular folding of the retina’s ILM and nerve fiber layer (NFL). In rare instances, significant vision loss can occur when further tractional forces result in cystoid macular edema (CME), macular holes, or retinal detachment (RD).
Etiology
ERMs have been described differently in the literature, depending upon their etiology, appearance, and/or clinical stage, and include idiopathic ERM, preretinal membrane, preretinal gliosis, preretinal macular fibrosis, cellophane maculopathy/retinopathy, crinkled cellophane, star folds, surface-wrinkling retinopathy, macular pucker, pigmented macular pucker, pigmented preretinal membrane, primary retinal folds, and silent central retinal vein (CRV) obstruction. Some ERMs may be associated with ocular and/or facial trauma, retinal vascular disorders (e.g., diabetes), vitreous hemorrhage, retinal breaks and detachment, intraocular inflammation (e.g., toxoplasmosis), photocoagulation, or previous anterior segment or vitreoretinal surgery. Most IERMs are idiopathic.
The Patient
Symptoms and signs of IERMs are dependent upon the extent of the disease process and the presence of secondary tractional sequelae, such as CME.
Clinical Symptoms
Typically, there are no subjective complaints, and IERMs are found on routine examination. However, patients may complain of blurred vision and/or metamorphopsia. Decreased depth perception, diplopia, and absolute scotoma are also infrequently observed.
Clinical Signs
See Figure 18-3.
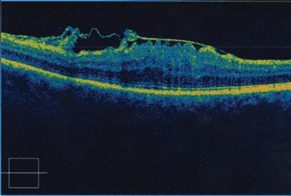
Figure 18-3. OCT demonstrating large ERM.
Prominent Signs
- Decreased visual acuity (dependent upon the extent and location of membrane formation and presence of secondary sequelae)
- Amsler grid metamorphopsia
- Loss of foveal reflex
- Semitranslucent to opaque gathered-pleat or volcano pattern appearance of IERM with deviation of retinal vasculature and retinal folds
Subtle Signs
- Cellophane reflex of retinal surface (best observed with the binocular indirect ophthalmoscope, accessory biomicroscopic lenses, and/or red-free filter of the direct ophthalmoscope)
- Perimacular vasculature may be slightly tortuous, and vessels may appear to straighten toward the area of the IERM
Other signs include PVD (“partial” versus “complete”), retinal thickening (suggesting macular edema), macular hole, and retinal break/detachment.
- IERM is much more prevalent in patients over the age of 50 (6.4% of individuals within this age group are thought to have the condition).
- Unilateral IERM occurs in 70% to 90% of cases.
- Approximately 1% to 2% of unilateral cases eventually become bilateral.
- IERM affects both sexes equally.
- PVD occurs in 80% to 90% of cases.
- CME has been reported to occur in 16% of patients with IERM.
- In 77% of cases, vision is 20/60 or better, while 61% have acuity of 20/40 or better.
Significant History
- Usually none (possibly PVD)
Ancillary Tests
Ocular assessment of IERM would include best-corrected visual acuity, pupil assessment, Amsler grid, color vision, dilated fundus examination, and optical coherence tomography (OCT).
Fluorescein angiography (FA) is usually not necessary but may be indicated when acuity is worse than 20/40 (especially when a partial PVD is present), concurrent CME is suspected, or the condition occurs in younger individuals.
The Treatment
In most cases, surgical intervention is not warranted as IERMs are usually self-limiting with a final visual acuity of 20/40 or better. Patients are instructed on routine Amsler grid usage, and follow-up examinations are scheduled according to individual case severity.
Historically, surgery was considered only when visual acuity was 20/60 or worse and after contemplating preexisting permanent macular damage as well as anticipated surgical trauma. More recently, earlier membrane removal has been advocated for cases in which OCT demonstrates a disrupted photoreceptor integrity line. When surgery is performed, a pars plana approach with a closed intraocular microsurgical technique (vitrectomy with membrane peeling) is employed. Stabilization of visual acuity typically occurs 1 to 3 months postoperatively and in 83% of cases improves by at least two Snellen lines. ERMs recur at an incidence of approximately 3% to 4%.
Ocular Histoplasmosis Syndrome
Michael J. Trad
ICD-9: 115.02
 THE DISEASE
THE DISEASE
Pathophysiology
Ocular histoplasmosis syndrome (OHS) presents initially in the posterior pole, peripapillary area, and peripheral retina as a bilateral, multifocal choroiditis. Active “histo” lesions are comprised of lymphocytes, plasma cells, and macrophages and may disrupt Bruch’s membrane and subsequently damage the retinal pigment epithelium (RPE) and outer retinal layers.
It is believed that 10 to 30 years after the initial infection, in which inhaled particles of fungus enter the body, reactivation of the disease and subretinal neovascular membrane (SRNVM) progression may occur. A potential visually devastating maculopathy may ensue when SRNVM extension results in hemorrhagic RPE and/or neurosensory retinal detachment (RD), and ultimately disciform scar formation.
Etiology
Systemic histoplasmosis is a fungal infection caused by the yeast Histoplasma capsulatum. H. capsulatum spores present in soil (in close proximity to the droppings from chickens, other birds, and bats) are inhaled into the pulmonary system and then travel via the bloodstream to the eye (choroid) and/or other organ systems.
Although cases of OHS may be found anywhere throughout the United States because of population migration, OHS typically affects individuals living in the so-called histo belt of the Ohio and middle Mississippi valleys, Alabama, Arkansas, Illinois, Indiana, Iowa, Kansas, Kentucky, Louisiana, Maryland, Mississippi, Missouri, Nebraska, Ohio, Oklahoma, Tennessee, Texas, West Virginia, and Virginia. OHS usually occurs in immunocompetent persons without overt systemic manifestations, but may present opportunistically in immunocompromised patients (e.g., AIDS).
The Patient
Clinical Symptoms
Patients are usually asymptomatic unless significant macular or peripapillary retinal involvement occurs. In these instances, blurred or distorted vision and field loss may be experienced.
Clinical Signs
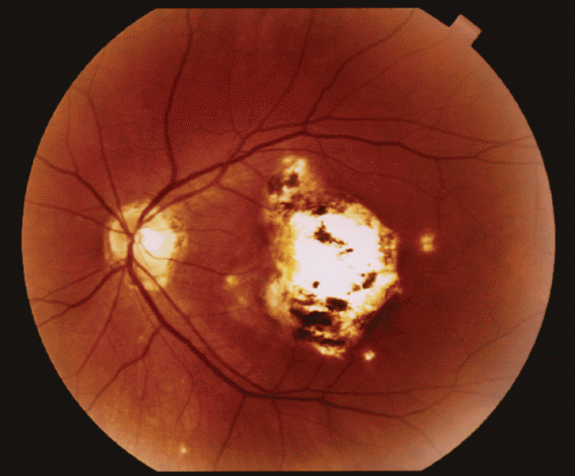
Figure 18-4. Macular scar and peripapillary atrophy in OHS.
Figure 18-5. Peripapillary atrophy and macular/perimacular chorioretinal scars in OHS.
Prominent Signs
- Classic triad of peripapillary atrophy, peripheral chorioretinal atrophic spots, and macular subretinal neovascularization
- Absence of anterior uveitis
- Disciform macular scar
- Metamorphopsia or relative scotoma on Amsler grid
- Hemorrhagic/serous retinal and RPE detachment
- Optic disc edema, choroidal hemorrhage
- Vitreous hemorrhage (infrequent)
Subtle Signs
- Absence of vitritis (vitritis concurrent with OHS-like signs may indicate an inflammatory disorder, such as pseudo-POHS, a subtype of acute zonal occult outer retinopathy)
- Gray-green area in macular or peripapillary area indicating the presence of an SRNVM
- Linear streak lesions at the equator, which run parallel to the oral serrata (infrequent)
Demographics
- OHS patients typically range in age from 20 to 50 years.
- OHS occurs six times more frequently in whites than in blacks.
- OHS occurs with equal frequency between the sexes.
- Bilateral macular involvement is more common in men than in women.
- Maculopathy presents in individuals between the ages of 20 to 40 years.
- Each year in the United States, at least 2,000 individuals experience significant visual loss because of OHS.
- Bilateral macular involvement is more common in men than in women.
Significant History
Past or present residence in the histo belt; past exposure to chickens, pigeons, or parakeets; immunodeficiency states (e.g., AIDS may reactivate OHS); stress may precipitate macular involvement.
Ancillary Tests
Ocular assessment for OHS may include visual acuity, color vision, Amsler grid, confrontation fields, pupils, biomicroscopy, tonometry, dilated funduscopy, fundus photography, and optical coherence tomography (OCT).
Laboratory studies are rarely necessary, as the diagnosis is usually made on fundus appearance alone. However, in rare instances, chest x-ray and HLA gene typing (DRw2 and B7 genes) may be utilized.
Fluorescein angiography (FA), indocyanine green (ICG) angiography, and OCT may be useful in cases of presumed or active maculopathy.
The Treatment
The Verteporfin in Ocular Histoplasmosis Study demonstrated efficacy for photodynamic therapy (PDT) of SRNVMs, and subsequently PDT has been utilized for subfoveal, juxtafoveal, extrafoveal, and peripapillary choroidal neovascular membranes (CNVMs). Anti-VEGF therapy has also been successful.
Recurrence of SRNVMs necessitates home Amsler grid testing, monitoring of systemic hypertension (HTN) (which may exacerbate recurrences), and yearly ocular examination (or more frequently if necessary).
Ocular Toxoplasmosis
Nicky R. Holdeman
ICD-9: 771.2—CONGENITAL TOXOPLASMOSIS
ICD-9: 130.0—ACQUIRED TOXOPLASMOSIS
 THE DISEASE
THE DISEASE
Pathophysiology
Toxoplasma gondii is the most common cause of infectious retinochoroiditis in the world, accounting for 30% to 50% of cases in otherwise healthy individuals. In addition to retinochoroiditis, the organism may also produce other functional changes including a dense vitritis, vitreous detachment, iridocyclitis, perivasculitis, retinal detachment (RD), neovascularization, cataracts, and glaucoma.
Recurrent retinochoroiditis occurs when tissue cysts release parasites that invade and destroy retinal cells. Since the organism is an intracellular parasite, the retina sustains the primary insult and the major damage. As they proliferate, parasites stimulate inflammatory reactions and the expression of vascular endothelial growth factor (VEGF), resulting in clinically apparent lesions. Factors related to reactivation of disease in otherwise healthy individuals are not known and recurrences cannot be predicted.
Etiology
T. gondii is a small obligate intracellular protozoan parasite affecting humans and animals; nearly one third of humanity has been exposed to this parasite. Multiplication occurs in the intestines of cats and the oocyst forms are shed in the stool. Cysts may remain viable in the soil for extended periods.
The most common routes of human transmission are by intrauterine infection, by ingestion of contaminated food or undercooked meat, or through inhalation of oocysts in cat litter.
Although congenital infection can affect any organ, ocular disease is the most common manifestation resulting in irreversible damage to the retina in utero. Toxoplasma organisms invade the retina of the fetus, where they may change into the cystic form. Most serious, active cases are thought to represent a late manifestation of an intrauterine infection, where the cysts rupture and liberate parasites in surrounding cells.
The Patient
Clinical Symptoms
The patient will generally not complain of pain, but rather of an increase in floaters and a reduction of vision in the affected eye.
Clinical Signs
The appearance of a toxoplasmosis lesion varies depending on whether the infection is in an active or inactive state, and on the immune status of the patient.
Active retinochoroiditis, in an immunocompetent patient, typically appears as a unilateral, creamy-white, fluffy, necrotic, retinal lesion with blurred margins. Recurrent disease often occurs next to an old scar to produce a “satellite lesion.” Usually, a hazy vitreous overlies the lesion giving a “headlight in the fog” appearance (Fig. 18-6).
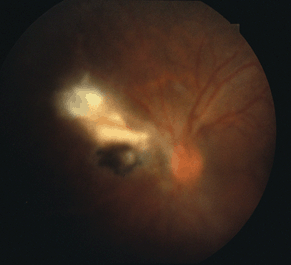
Figure 18-6. Reactivation of toxoplasmosis retinochoroiditis with overlying vitritis.
In immunocompromised patients, the active condition is bilateral in 20% of cases. The retinal lesions are often extensive and deep (punctuate outer retinal toxoplasmosis or PORT) and may consist of large areas of retinal necrosis or retinochoroiditis, without a prominent vitritis or adjacent preexisting retinal scars. These atypical retinal lesions may be associated with cerebral involvement, myocarditis, and pneumonitis.
The ocular complications of toxoplasmosis may include subretinal neovascular membranes (SRNVMs) (see Fig. 18-7), vessel occlusions, preretinal gliosis, retinal breaks, RDs, macular edema, papillitis, optic nerve atrophy, synechia, cataract, and/or glaucoma. A mild anterior chamber reaction may be seen in some patients.
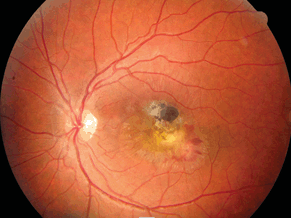
Figure 18-7. Toxoplasmosis lesion of the macula with subretinal bleeding.
Inactive lesions produce a well-demarcated chorioretinal scar with bare sclera often surrounded by hypertrophic retinal pigment epithelium (RPE).
Demographics
It has long been believed that the vast majority of cases of ocular toxoplasmosis involving adults are the late sequelae of a congenital infection, with the mother acquiring a primary infection during pregnancy and then passing it to the fetus transplacentally.
The frequency of ocular involvement in postnatally acquired toxoplasmosis is not known, but the incidence of acquired infections is probably much greater than originally suspected. There is clear evidence that acquired toxoplasmosis can induce ocular lesions; however, many of these acquired cases produce self-limited and benign lesions.
Congenital infection has been estimated to affect 3,000 infants born in the United States each year. When primary infection of the mother occurs during pregnancy, there is a 40% chance of fetal infection. As a rule, IgG antibodies to Toxoplasmosis protect the fetus, but that is not always the case.
Recurrences often manifest between ages 11 and 40, with the mean age of first presentation being approximately 29 years. The disease is rather uncommon in early childhood and after the age of 50, but severe or acquired cases of ocular toxoplasmosis have been reported in older patients without suspected or documented immunodeficiency.
Recurrences have been observed in 49% of patients within 3 years.
Legal blindness (BCVA ≤ 20/200) in at least one eye may be expected in 24% of patients (i.e., macular involvement, vitreous hemorrhage, SRNVM, or RD).
Significant History
- Is the patient aware of having contracted any congenital infections?
- Does the patient eat raw or undercooked lamb, pork, or chicken?
- Is there an exposure to cats or cat litter?
- Infected water, organ transplantation, and blood transfusions are potential sources of transmission, but are rare in the Unites States
- Inquire about immunosuppression or risk factors for AIDS in patients with active bilateral, atypical, or multifocal lesions.
Ancillary Tests
The diagnosis of ocular toxoplasmosis is usually made by the classic fundus findings; however, several tests can be used to confirm clinical impressions or assist in atypical cases.
- Serology is supportive but not diagnostic in adults, as 20% to 70% of the general population will show positive titers.
- The most commonly used serologic tests are the IFA and ELISA for toxoplasmosis which can be done on blood, cerebrospinal fluid (CSF), and aqueous. These tests permit separation of IgM and IgG antibodies. The presence of IgM antitoxin titers indicates a recent acquisition of infection. Recurrent retinochoroiditis is usually associated with stable, low IgG titers and no IgM antibody. A positive test, even at a 1:1 dilution, corroborates the diagnosis.
- In difficult or atypical cases, anterior chamber paracentesis to detect local antibody production may be helpful. If IgG antibodies in the aqueous are higher than in the serum, the diagnosis is supported. In addition, polymerase chain reaction tests to detect T. gondii DNA in the aqueous can help confirm the diagnosis and help differentiate ocular toxoplasmosis from similar retinal lesions caused by herpes virus or other infectious agents.
- The Sabin-Feldman dye test, which was the original standard test for toxoplasmosis, requires working with live Toxoplasma organisms and is seldom used today.
- Fluorescein angiography and optical coherence tomography may be useful in helping to identify subretinal neovascular membranes and/or macular edema.
The Treatment
The treatment for ocular toxoplasmosis is not uniform. Some lesions, if they are small and in the retinal periphery, are generally not a threat to vision, and if they occur in an immunocompetent host may simply be followed. Activity in the lesion may persist for up to 4 months, but it will spontaneously resolve.
Large lesions, or those that (a) threaten the optic nerve, macula, or a large retinal vessel (b) cause excessive hemorrhage, or (c) induce a significant inflammatory response, are often treated with a combination of systemic steroids plus one to three antitoxoplasmic agents. It should be noted, however, that there is a lack of evidence to support routine antibiotic treatment for acute toxoplasmic retinochoroiditis and that treatment may be associated with adverse effects.
Corticosteroids are rarely employed in the treatment of immunocompromised patients and should never be used without antimicrobial therapy. Steroids are often given concurrently with antitoxoplasmic drugs to alleviate inflammation, especially when the lesions are located near the macula or the optic nerve.
- Prednisone 40 to 80 mg p.o. q.d. began after the second or third day of antitoxoplasmic therapy until day 10 and then tapered gradually over a period of 4 to 5 weeks. Stop steroids before discontinuing antitoxoplasma medications.
Antitoxoplasmic drugs include the following:
- Pyrimethamine—loading dose of 100 to 200 mg p.o. the first day and then 25 mg p.o. b.i.d. for 3 to 6 weeks. Follow complete blood counts and give folinic acid (leucovorin) 5 mg p.o. three to four times weekly to minimize bone marrow depression (thrombocytopenia, leukopenia, megablastic anemia). Continue leucovorin for 1 week after stopping pyrimethamine.
- Sulfadiazine—loading dose of 2 to 4 g p.o. the first day, then 1 g p.o. q.i.d. for 3 to 6 weeks depending on clinical response.
- Clindamycin—150 to 300 mg p.o. q.i.d. Useful adjunct to above therapy or may be used with only sulfadiazine. If the patient has a sulfa allergy, clindamycin may be used with pyrimethamine. Clindamycin concentrates in the choriod but is expensive. Monitor for pseudomembranous colitis. Limit treatment to 4 weeks.
- Azithromycin—500 mg on day 1 followed by 250 mg/d, alone or in combination with pyrimethamine, appears to be effective in treating sight-threatening lesions with fewer side effects than other antibiotics.
- Tetracycline—loading dose of 1 g p.o. on the first day, then 500 mg p.o. q.i.d. for 3 to 6 weeks.
- Trimethoprim (160 mg)/sulfamethoxazole (800 mg)—1 tablet p.o. b.i.d., with or without clindamycin. Many uveitis specialists concur that TMP/SMX is an effective therapy and is generally well tolerated. There is also some evidence that long-term treatment (every 3 days for up to 20 months) with TMP/SMX significantly reduces the rate of recurrence of retinochoroiditis in patients with a history of multiple previous recurrences.
Note: Since some of these drugs may have significant side effects, it is advisable to follow these patients with an internist.
Success has been reported with intravitreal injections of clindamycin and dexamethasone and may offer an additional treatment strategy in patients who are unable to afford or tolerate systemic therapy, or whose disease progresses despite systemic therapy.
Intravitreal injection of bevacizumab (Avastin) appears to be an effective and safe treatment in patients with choroidal neovascularization (CNV) secondary to ocular toxoplasmosis.
There appears to be an increased risk of reactivations of ocular toxoplasmosis following cataract extraction. This finding would imply that prophylactic treatment with antitoxoplasmic agents prior to and after surgery might be prudent for patients at risk for visual loss. Consider TMP/SMX p.o. b.i.d.
Anterior segment inflammation varies from a minimal response to a severe granulomatous iritis and is treated with topical cycloplegics (e.g., atropine 1% b.i.d.–t.i.d.) and/or topical steroids (e.g., prednisolone acetate 1% q.i.d.) as the need exists. Periocular corticosteroid injections can cause dissemination and should not be given.
Toxocariasis
Nicky R. Holdeman
ICD-9: 128.0
 THE DISEASE
THE DISEASE
Pathophysiology
Toxocariasis is a clinical syndrome resulting from parasitic invasion of human viscera with subsequent migration of the larvae to various organs. Most ocular damage results from the inflammatory response that occurs following death of the larva. Toxocara does not develop beyond the larval state in the incidental human host, and thus eggs are not found in the stool of humans.
Etiology
Toxocariasis most commonly results from infections with larvae of the canine roundworm, Toxocara canis. The adult worms live in the intestinal tracts of their primary hosts and release large numbers of eggs in the stool. The eggs can remain active and infective in the soil for months to years.
Human infections are generally acquired by young children who ingest the ova from soil or sand (geophagia) contaminated with animal feces, most often puppies. The embryonated eggs can also be acquired from contaminated raw vegetables or infected raw meat. The eggs hatch in the intestine and liberated larvae then penetrate the mucosal wall and are disseminated by the systemic circulation to the liver, lung, brain, kidney, striated muscle, central nervous system (CNS), heart, and the eye where they may produce an eosinophilic granulomatous inflammation. Remains of larvae are found within the granulomas, which contain eosinophils and histiocytes.
The Patient
Clinical Symptoms
Symptoms will vary depending on the organ involved. Children, usually before the age of 3, may develop acute systemic disease (i.e., visceral larvae migrans [VLM]). If so, the migrating larvae may produce symptoms such as malaise, fever, cough, skin rash, and/or abdominal pain.
Patients with ocular toxocariasis are usually older children or young adults, who complain of visual impairment in one eye, or may be referred for evaluation of leukocaria or strabismus. Eye pain is not typical in this condition.
Clinical Signs
Patients with visceral larva migrans may demonstrate lymphadenopathy, hepatomegaly, fever, wheezing, or a variety of other findings with diverse organ invasion.
In the eye, toxocariasis can have the following three different presentations:
- Peripheral chorioretinal granuloma (~50% of patients)—usually associated with dense fibrous bands in the vitreous with dragging of the macula; may mimic pars planitis.
- Posterior pole chorioretinal granuloma (~25% to 50% of patients)—a white mass, 1 to 6 mm in diameter, is seen with an accompanying vitritis; usually located in the macula or in the papillo-macular bundle (Fig. 18-8).
- Endophthalmitis (<25% of patients)—chronic, diffuse, painless uveitis with a cloudy vitreous. There may be a secondary cataract, cyclitic membrane, and/or RD.
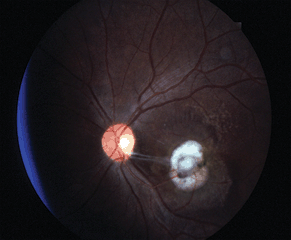
Figure 18-8. Toxocara posterior pole granuloma with fibrous vitreous bands extending from the lesion.
Note: It is rare to have VLM and ocular disease concurrently.
Ocular toxocariasis is typically a unilateral disorder, but confirmed cases of bilateral panuveitis have been reported.
Demographics
- The overall prevalence of Toxocara infection of the eye is about 1% of the general uveitis population.
- Visceral larva migrans usually presents around the age of 2 years.
- Most cases of ocular toxocariasis occur in children 5 to 10 years of age, with a range of 2 to 30 years. By the time the child seeks eye care, systemic manifestations have usually resolved.
- The largest number of cases of ocular toxocariasis has been reported in the United States; however, the disease has been seen in many countries worldwide.
Significant History
- Is there known exposure to puppies, especially 2 weeks to 6 months of age? (>50% of puppies and 20% of adult dogs harbor the parasite)
- Is there a history of geophagia?
- Did the child play in a sand box?
- Is there a history of ingesting uncooked foods?
Ancillary Tests
The diagnosis of both visceral larva migrans and ocular toxocariasis is typically made based on the appropriate signs and symptoms, together with a history of exposure to infected pets or pica. In atypical cases, the diagnosis can be aided by an ELISA titer of undiluted serum, which has a high sensitivity and specificity (90%) for toxocara antibodies.
Occasionally, in cases of ocular toxocariasis, the serum ELISA titers may be normal but antibody titers of intraocular fluids may be elevated which permits a diagnosis. Unfortunately, ELISA analysis of intraocular fluids may only be available at a few isolated facilities.
Visceral larva migrans is often associated with a marked leucocytosis, with 30% to 80% due to eosinophils. Ocular toxocariasis is usually not associated with a peripheral eosinophilia; however, eosinophils found in the vitreous can be helpful in the diagnosis.
Ultrasonography, to detect the intraocular calcifications of a retinoblastoma, can assist in the differential diagnosis. B-scan can also be helpful when vitritis or other opacities preclude a view of the fundus.
Optical coherence tomography can image the posterior pole granulomas and show factors contributing to vision loss such as intraretinal or subretinal fluid.
The Treatment
The medical management of ocular toxocariasis is unclear as no proven treatment is available. While the acute systemic infection is sometimes treated with thiabendazole, mebendazole, albendazole, or diethylcarbamazine, the use of these antihelminthic agents is of questionable benefit in ocular toxocariasis as the disease is self-limiting, the amount of ocular absorption is unknown, and the reaction to a dead nematode can increase ocular inflammation. If oral antihelmenthics are employed, it is recommended that they be used in conjunction with steroids to decrease the inflammatory process.
Suggested regimens for ocular toxocariasis may include a combination of the following:
- albendazole 400 mg p.o. b.i.d. for 5 days. Use is controversial
- prednisone 30 to 60 mg p.o. q.d. for 5 to 10 days followed by taper to control the vitreitis (in some cases, periocular injections of steroids may be preferable to oral usage).
Anterior uveitis, if present, is typically mild and should be treated with topical steroids and cycloplegics as clinically indicated.
If subretinal larva is visualized outside the foveal area, photocoagulation or cryocoagulation may be used to destroy the organisms.
Surgery is sometimes required for complications such as tractional retinal detachments.
Note: Controlling toxocara infections in dogs and puppies is essential if there are children in the household. Complete cooking of meats, thorough rinsing of fruits and vegetables, and hand washing are important measures for preventing the human infection.
Age-Related Macular Degeneration
Nakhleh E. Abu-Yaghi, Helmut Buettner, Sophie J. Bakri
ICD-9: 362.50
 THE DISEASE
THE DISEASE
Pathophysiology
As the macula ages, many ultrastructural changes occur: the photoreceptors are reduced in density and distribution, residual bodies accumulate in the retinal pigment epithelium (RPE), basal laminar deposits form in Bruch’s membrane, and the choriocapillaris starts to involute. Age-related macular degeneration (AMD) is a distinct process that is characterized by specific non-neovascular and neovascular abnormalities.
Drusen are the hallmark of non-neovascular AMD. They are small round yellowish lesions corresponding to abnormal thickening of the inner aspect of Bruch’s membrane. Histologically, granular lipid rich materials with widely spaced collagen fibers form basal laminar deposits, whereas phospholipid vesicles and electron-dense granules form basal linear deposits within the inner aspect of Bruch’s membrane. This thickened Bruch’s inner layer, together with the RPE, can separate from the rest of Bruch’s membrane to form a pigment epithelial detachment.
Other non-neovascular abnormalities in the RPE include contiguous areas of RPE loss or attenuation, termed geographic atrophy. This is associated with loss of the overlying photoreceptors. Focal hyperpigmentation of the RPE is another non-neovascular change and is a risk factor for progression to the more advanced forms of AMD.
All non-neovascular changes increase the chance of a break occurring in Bruch’s membrane, allowing neovascular tissue from the choriocapillaris to perforate the outer aspect of Bruch’s membrane. This is neovascular or exudative AMD. Fibrovascular proliferation can occur, disrupting the normal architecture of the outer retina. A disciform scar can eventually form.
Etiology
AMD is a complex disease caused by intricate interactions of environmental factors and genetic predisposition. Many theories were postulated to understand the etiology of AMD. An inflammatory model was supported by genetic association studies which revealed allelic variants of genes coding the alternate complement pathway, particularly compliment factor H. Two genetic mutations, namely Tyr402HIS and Ala69Ser (both on the long arm of chromosome 10), may explain 75% of the genetic risk of AMD. These and other mutations have been clearly validated in white populations but do not seem to confer additional risks in other races. Other factors that predispose to AMD include systemic hypertension, cardiovascular disease, and smoking. Exposure to sunlight (ultraviolet light [UV]) was thought to play a role in the disease process, but no causal relationship was established.
The Patient
Clinical Symptoms
- Initially may be asymptomatic
- Blurred vision
- Metamorphopsia
- Central blind spot
Clinical Signs
The hallmark clinically detectable abnormality and earliest sign of AMD is round, slightly elevated yellowish-gray deposits under the RPE primarily in the posterior pole, which are called “drusen” (Fig. 18-9). Drusen, which are composed of eosinophilic material between the basement membrane of the RPE and Bruch’s membrane, may become visible ophthalmoscopically as early as the fourth decade of life, and generally grow in number, enlarge with progressing age, and may become confluent. Mottled pigment eventually accumulates around the drusen. At this stage, visual acuity is usually normal, but symptoms of requiring more light to read or mild metamorphopsia may be noted. This manifestation of AMD is also referred to as the dry form of macular degeneration and is seen in about 90% of individuals with AMD. Atrophy of the RPE may develop in a sharply outlined or geographic pattern (geographic atrophy), which, when involving the fovea, leads to severe loss of central vision.
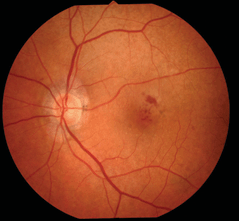
Figure 18-9. Age-related macular degeneration. (Photo courtesy of Leonid Skorin Jr.)
The individual with large drusen surrounded by pigment proliferation is at greatest risk of developing the wet or exudative form of AMD encountered in about 10% of patients with the disease. The wet form of AMD is characterized by leakage of fluid alone, resulting in a pigment epithelial detachment and/or serous neurosensory retinal detachment (RD), or leakage of fluid and blood from neovascularization arising from the choroidal circulation. The neovascularization, if left untreated, eventually fibroses to form scar tissue (disciform scar), which can extend under the entire posterior pole. The retinal photoreceptor cells overlying such a scar degenerate, resulting in severe loss of central vision, also noted by the patient as a blurred spot or scotoma in the central visual field.
Demographics
Age-related macular degeneration is the most common cause of irreversible legal blindness in the United States and Western Europe in patients over 60 years of age. Ten percent of the population over age 65 in the United States exhibit fundus changes of AMD (large drusen and pigment mottling in the macula), and it is currently estimated that 1% will be legally blind (20/200 or less vision bilaterally) from the disorder. AMD affects primarily blue-eyed, white individuals, women more often than men. It rarely causes visual loss in African Americans. Many individuals with AMD have other family members affected by the disease.
Ancillary Tests
In addition to a detailed eye examination including biomicroscopy, a number of tests are extremely helpful in diagnosing and managing patients with AMD. The Amsler grid allows the detection and localization of changes in the central visual field caused by AMD. It also allows the patient with AMD to self-monitor the central visual field for the development of visual symptoms or their changes. FA is extremely helpful in the identification and classification of anatomic and functional abnormalities associated with AMD. Occasionally, indocyanine green angiography used as an adjunct can be helpful in visualizing the choroidal neovascular membrane (CNV) when it is obscured by blood. These angiographic techniques are particularly useful in determining the location and size of the CNV, important for the planning and performance of their treatment. Optical coherence tomography (OCT) can provide cross-sectional high-resolution images of the retina, pigment epithelium, and inner choroid in the macular area, helpful in diagnosing and monitoring the disease or its response to treatment.
The Treatment
Population-based studies have shown that individuals who have consumed a diet rich in antioxidants (green leafy vegetables, red wine) throughout their lives have a lower risk of developing and losing vision from AMD. These studies led to a large, prospective randomized trial of the effect of high-dose vitamins and minerals on the progression of AMD in patients with large drusen in the macula. The Age-Related Eye Disease Study (AREDS) supplements consists of daily doses of vitamin A (28,000 units), vitamin C (500 mg), vitamin E (400 IU), zinc (80 mg), and copper (2 mg). In the AREDS, individuals who received these vitamins and mineral supplement had a lower risk of both progression of their AMD and visual loss from AMD as compared to individuals who received a placebo. Other measures aimed at preventing development or progression of AMD include cessation of smoking, optimal control of cardiovascular disease and risk factors, and minimizing exposure to UV by wearing appropriate UV-blocking glasses. The AREDS 2 study is currently evaluating the effects of high supplemental doses of dietary xanthophylls (lutein and zeaxanthin) and omega-3 long-chain polyunsaturated fatty acids on the development and progression of AMD. In this study, beta carotene (vitamin A) has been eliminated from the original AREDS formulation, and the dose of zinc has been reduced from 80 to 25 mg.
Several ongoing clinical trials are looking for treatment options for geographic atrophy. One example is the complement inhibitors, given systemically or intravitreally the evolution to atrophic AMD.
Treatment of Exudative AMD
Historically, laser photocoagulation has been used to treat well-demarcated CNVs. The Macular Photocoagulation Study showed that after 5 years of randomization, the proportion of eyes, treated with laser or not, that maintained vision within 1.5 lines of the initial management was very small. Laser photocoagulation is still used sometimes for extrafoveal and peripapillary CNVs, although recent advances in pharmacotherapy have considerably limited its use.
Photodynamic therapy (PDT), a combination of the photosensitizing drug verteporfin and infrared laser irradiation, was evaluated in many studies to treat exudative AMD. A large randomized collaborative study (Treatment of Age-Related Macular Degeneration with Photodynamic Therapy) showed that 67% of eyes with predominantly classic subfoveal neovascularization treated with verteporfin had lost fewer than 15 letters of vision after 12 months as compared to only 39% of eyes treated with placebo. The verteporfin treatment benefit was still present after 2 years. The verteporfin in photodynamic therapy trial evaluated treatment of subfoveal occult CNVs. At 2 years, 45% of treated eyes lost fewer than 15 letters compared to 32% of sham-treated eyes. These studies showed that PDT reduces the risk of further loss in vision by around 50%. In the pre–anti-VEGF (vascular endothelial growth factor) era, PDT was an appropriate treatment for subfoveal CNV, new or recurrent, in predominantly classic lesions, or occult lesions with a visual acuity of 20/50 or worse, or with a visual acuity of better than 20/50 with a lesion size less than a disc diameter.
The pharmacologic inhibition of VEGF is the mainstay of treatment in wet AMD currently. Ranibizumab (Lucentis) is a recombinantly produced, humanized antibody (Fab) fragment that binds VEGF and is FDA-approved to treat wet AMD as an intravitreal injection. Studies have shown that 95% of ranibizumab-treated individuals experienced visual improvement or stabilization compared to 62% of sham-treated patients after 12 months, with 40% of treated patients achieving improvement by 15 letters or more. Multiple re-treatments are necessary, sometimes at monthly intervals for several years, and this can be guided by both clinical and imaging parameters. Bevacizumab (Avastin) is a full-length monoclonal antibody against VEGF approved for metastatic colorectal cancer, but it is widely used off-label to treat wet AMD. A randomized, multicenter, clinical trial, the CATT trial (Comparison of ARMD Treatments Trials), is designed to compare the efficacy and safety of bevacizumab and ranibizumab.
Combination treatment is a tempting option for many reasons. The complexity of the disease calls for a treatment approach that offers synergistic modes of action to target the multiple components of AMD and to reduce the frequency of re-treatments and stabilize the visual improvement. The FOCUS study and the PROTECT trial evaluated the safety and efficacy of ranibizumab in combination with PDT versus PDT alone. At 1 year, 91% of treated subjects had stable vision compared with 68% under PDT alone. The number of PDT sessions was 2.3 in the combination subjects versus 3.4 in monotherapy. Similar results were reported from two nonrandomized studies that evaluated the combination of bevacizumab and PDT. Combining intravitreal steroids with PDT and/or anti-VEGF treatments is also an attractive option. Corticosteroids inhibit VEGF expression, vascular permeability, and inflammation, and using them in combination appears to be safe, especially in pseudophakic patients.
Many surgical approaches have tackled AMD. The Sub-macular Surgery Trials showed that surgical removal of subfoveal CNV compared to observation did not improve vision. Macular translocation, a surgical procedure to relocate the macular retina over healthier RPE adjacent to the neovascularization, is only of historic/academic interest now and is very rarely performed in the era of anti-VEGF therapy. Transplantation of RPE-like cells from embryonic stem cells may represent the future of surgical approaches to AMD.
Low vision rehabilitation remains an important part of advanced AMD management and should be offered to all patients with bilateral advanced disease.
Stargardt’s Disease
Jerome Sherman
ICD-9: 362.75
 THE DISEASE
THE DISEASE
Stargardt’s disease or dystrophy, occasionally termed juvenile macular dystrophy, is the most common form of inherited juvenile macular degeneration. It is characterized by progressive and symmetrical atrophy of the macula that affects individuals with previously normal vision. Stargardt’s disease is often considered a form of fundus flavimaculatus, where macular atrophy predominates early in the disease progression. Perhaps, the most accurate name is “Stargardt’s Disease with Fundus Flavimaculatus” because most patients have both the macular atrophy and the characteristic flecks.
Pathophysiology
Although most eye clinicians regard Stargardt’s disease and fundus flavimaculatus as different manifestations of the same disease, for classification purposes, they are interchangeable. The term fundus flavimaculatus is applied when the characteristic subretinal flecks, which are fish-tail or pisciform in shape, are scattered throughout the fundus. When the flecks are very subtle, confined to the posterior pole or nonexistent, the macular atrophy is properly termed Stargardt dystrophy. Although some patients present initially with only the macular atrophy while others present with only the flecks, long-term follow-up of either group reveals the eventual ophthalmoscopic finding of both the macular atrophy and the characteristic flecks.
Stargardt’s disease has its onset within the first two decades of life. A patient initially presents with a decrease in visual acuity. Although visual acuity loss is often proportional to the degree of macular atrophy, it is not at all rare for a patient to present with reduced visual acuity but with a normal appearing fundus. Numerous patients with an eventual diagnosis of Stargardt’s disease have been initially misdiagnosed as malingering. Macular lesions may precede the flecks. Thus, the diagnosis of Stargardt’s disease is sometimes made before other signs of fundus flavimaculatus are evident.
Etiology
Stargardt’s disease is most typically a result of an autosomal recessive gene. As with all autorecessive disorders, a high incidence of consanguinity can be established through the appropriate questions. A very small number of families have an autosomal dominant form. All autosomal recessive forms of Stargardt’s and fundus flavimaculatus have been mapped to the short arm of chromosome 1. The autosomal dominant form of these disorders map to chromosome 13q34 and the long arm of chromosome 6. It has been found that the causal gene of Stargardt’s disease, ABCA4 (located on the short arm of chromosome 1), is a mutated photoreceptor cell-specific adenosine triphosphate–binding transporter. It is expressed in rod but not cone photoreceptors. The variations in sequencing of the ABCA4 gene appear to be responsible for the varied clinical presentations observed in patients with Stargardt’s dystrophy.
The Patient
Clinical Symptoms
The initial presenting symptom is always that of decreased central vision in an individual with prior normal vision. In the later stages of the disease, there may also be noticeable color vision defects. Night blindness is usually not a complaint. Photophobia is rare as an early complaint, but some patients report that they are bothered by glare.
Clinical Signs
The onset of Stargardt’s disease is usually in the first or second decade of an individual’s life. The most characteristic finding of Stargardt dystrophy is a progressive and symmetrical atrophy of the macula with previously normal vision. Visual acuity initially varies from 20/25 to 20/60 and progresses to 20/200. Although the progression of visual loss is variable, it is generally found to be similar with members of the same pedigree.
The initial fundus examination may be unremarkable with perhaps only a loss of the foveal reflex. Through the course of disease progression, a number of discrete yellowish pisciform flecks are evident at the level of the retinal pigment epithelium (RPE) surrounding the macula or are scattered throughout the posterior pole. Vessels are not usually attenuated. Over time, the RPE may take on a “bull’s-eye pattern” of loss. In the later stages of disease, the macula is often described as having a “beaten bronze” appearance with a horizontal oval area (~2 DD by 1.5 DD) (Fig. 18-10). As the macular atrophy continues to develop, the flecks extend to the midperiphery. There is an enlargement of the RPE cells that is thought to be secondary to excessive lipofuscin deposition in the zones of the flecks with a total disappearance of the cones, rods, and RPE cells in the circumfoveal zone. These small, deep flecks that appear around the lesion and in the periphery have the appearance of classic fundus flavimaculatus and thus are thought to be the same disease. It is when the macular lesions precede the flecks that the disorder may be diagnosed as Stargardt’s disease.
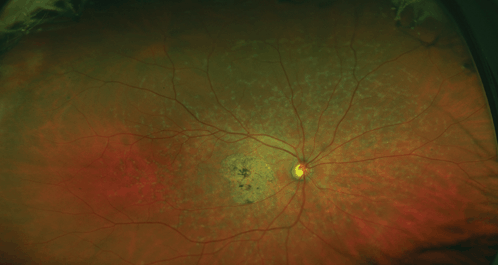
Figure 18-10. Stargardt’s disease (SD). A 41-year-old Hispanic female has vision reduced to 10/400. The Optomap plus with ResMaxTM image reveals a large “beaten bronze” maculopathy as well as widespread fundus flavi spots commensurate with Stargardt’s disease with fundus flavimaculatus. Unrelated to SD, the patient also has primary open-angle glaucoma.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


