Fig. 16.1
Pigment dispersion syndrome with Krukenberg spindle
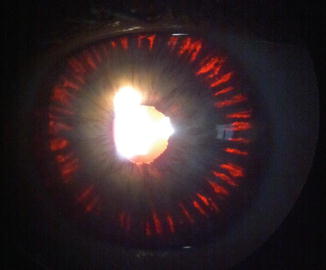
Fig. 16.2
Mid-peripheral, radial, slit-like pattern transillumination defects are seen most commonly inferonasally in young PDS/PG patients
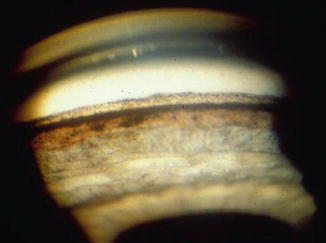
Fig. 16.3
In PDS, the angle is characteristically wide open, with a homogeneous, dense hyperpigmented band on the trabecular meshwork. The iris insertion is posterior and the peripheral iris approach is often concave
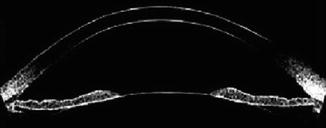
Fig. 16.4
Slit-lamp optical coherence tomography demonstrates iris concavity in a young PDS patient
Pathophysiology of Pigment Dispersion Syndrome
The underlying mechanism responsible for PDS is a concave iris contour that allows apposition of its posterior surface to the zonular bundles. The iris is also larger in patients with PDS, contributing to its concavity. Campbell hypothesized that friction between zonules and the peripheral iris in predisposed eyes is the cause of the pigment liberation in PDS [2]. Furthermore, a reverse pupillary block mechanism may exist, in which the iris drapes over the lens and acts as a “flap valve,” preventing aqueous in the anterior chamber from returning to the posterior chamber [3].
The act of blinking generates reverse pupillary block by compressing the anterior chamber and pushing the iris posteriorly [4]. When ultrasound biomicroscopy is performed on patients with untreated PDS, the iris configuration is initially concave. After an eyelid speculum is placed, inhibiting blinking, the iris gradually becomes less concave. As aqueous humor is produced, pressure builds in the posterior chamber pushing the iris forward. Once blinking is restored, the original concave configuration is reestablished. When the iris is pushed posteriorly by blinking, the aqueous humor in the posterior chamber moves into the anterior chamber. The pressure in the anterior chamber then exceeds that of the posterior chamber, but the aqueous humor in the anterior chamber cannot return to the posterior chamber because of the flap valve function of the iris, maintaining a concave configuration of the iris and forcing the iris pigment epithelium into contact with the zonular bundles. Mechanical rubbing during pupillary movement disrupts the iris pigment epithelium, releasing pigment granules into the aqueous humor. Greater iridozonular contact will cause greater pigment dispersion [5]. Trabecular endothelial cells phagocytose and remove pigment granules accumulated in the intertrabecular spaces [6–8]. When the amount of pigment granule accumulation exceeds the phagocytosis capacity of trabecular endothelial cells, the resistance of aqueous egress increases, elevating IOP.
Exercise (jogging, playing basketball, and bouncing during dancing) can cause the release of pigment as a result of pupillary movement in young PDS patients. We have seen pigment liberation and elevated IOP after exercise in a patient with PDS that was high enough to produce corneal edema. The type of exercise that induces pigment liberation may differ between patients. For example, we recall one patient who was a soccer player with PG. We asked him to return to our office after a workout that included running and kicking, and his pressures were normal after this exercise. However, when he returned to our office after a soccer match, his pressures were elevated. We concluded that the act of “heading” the ball (unique to the soccer match) was responsible for pigment liberation in his particular case. The phenomenon of exercise-induced IOP elevation in PDS can be prevented completely by miotic-induced relative pupillary block [9]. The use of laser iridotomy (discussed later) to eliminate reverse pupillary block inhibits exercise-induced pigment release incompletely [10].
Pharmacologic pupillary dilation in PDS may result in significant pigment liberation into the anterior chamber [11]. This pigment liberation may be accompanied by IOP elevation because of acute obstruction of the aqueous outflow pathway by pigment granules. However, the fact that delayed IOP elevation follows dilation in PDS is not well recognized, the astute clinician will measure IOP after dilation in all patients. In PDS, the maximal anterior chamber pigment dispersion may occur shortly after dilation, while the maximal elevation in IOP may occur several hours after dilation, when anterior chamber pigment is decreasing. This lag probably reflects the time necessary for pigment to obstruct the trabecular meshwork, reduce aqueous humor outflow, and for pressure to build within the eye.
Presentation
Patients with PDS are usually myopic, so fortunately they often seek eye care for spectacle or contact lens correction. Upon examination, they may be noted to have elevated IOP or pigment dispersion. PDS itself may present symptomatically. The classic presentation is that of a young (20- to 40-year-old) myopic male who experiences blurry vision or eye pain after exercise [12]. Occasionally, trauma followed by eye pain may be the heralding event, and furthermore trauma may exacerbate preexisting PG.
Examination
The key to making the diagnosis of PDS lies in performing a thorough exam with an appropriately high index of suspicion. With respect to refractive error, most patients with PDS will be myopic, some will be emmetropic, and very few will be hyperopic [13, 14]. On pupillary examination, hyperplasia or hypoplasia of the iris dilator muscle in PDS can cause deformation of the pupil in the direction of maximal iris transillumination. These changes may also result in anisocoria, with the less-involved pupil being smaller [15, 16]. In severe cases, an efferent pupillary defect may be present.
Slit-lamp examination should be performed with high magnification and in complete darkness with all room lights and computer monitors turned off to aid in the detection of anterior chamber pigment and iris transillumination defects. Corneal endothelial pigment appears as a central, vertical, brown band (Krukenberg spindle, with a “pine tree” shape), the pattern of pigmentation resulting from aqueous convection currents [17]. While the Krukenberg spindle may appear dense on slit-lamp biomicroscopy, this pigment almost never interferes with visual acuity, and, in fact, most of our younger patients with PDS (including those with dense Krukenberg spindles) usually have 20/15 corrected visual acuity. When the Krukenberg spindle is less dense, the endothelial pigment granules have the appearance of an extremely fine cinnamon powder and may be difficult to detect, particularly in patients who have passed the active liberation stage. Clinically, loss of iris pigment appears as a mid-peripheral, radial, slit-like pattern of transillumination defects seen most easily by retroillumination [18]. These defects are most often present inferonasally and are more apparent in lighter irides. The presence, amount, and distribution of iris transillumination defects should be noted as this information is useful for grading, for staging, and for following the disease.
Gonioscopy plays a pivotal role in the evaluation of patients with PDS/PG. On gonioscopy, the angle is typically wide open, the iris is inserted posteriorly into the ciliary body, and the configuration of the peripheral iris is concave. The amount of trabecular meshwork pigmentation should be graded and documented separately in the superior and inferior angles of both eyes. Pigment may also be deposited on Schwalbe’s line, on the zonules, on the posterior capsule of the lens, at the level of the insertion of the posterior zonular fibers (Zentmayer ring), and on the posterior lens central to Weigert’s ligament (Scheie’s stripe). Anterior segment imaging devices such as ultrasound biomicroscopy and anterior segment optical coherence tomography are useful for documenting the iris and angle configurations and comparing them before and after laser iridotomy.
After pupillary dilation, the anterior chamber should be evaluated in complete darkness for pigment and the IOP should be monitored. Ideally, the pressure is checked at least 1 h after dilation is complete. Treatment with a single drop of brimonidine is usually effective in treating pigment-related IOP spikes in the office. The peripheral retina must be carefully examined, as lattice degeneration may be present in up to 20 % of patients [19]. Furthermore, retinal breaks are present in up to 11.7 %, and rhegmatogenous retinal detachments requiring surgery may occur in 3.3 % of patients [19]. Optic nerve examination with careful drawing or stereophotography should be performed as for any patient, noting the size of the optic nerve, the presence or absence of peripapillary atrophy, nerve fiber layer defects, disc hemorrhages, or neuroretinal rim narrowing.
Diagnosis and Differential Diagnosis of PDS
To make the diagnosis of PDS, we require the presence of Krukenberg spindles, dense homogenous trabecular meshwork pigmentation, and a posterior iris insertion, with the presence of iris transillumination defects being confirmatory but not necessary. Many conditions other than PDS will cause the dispersion of pigment or of pigmented cells throughout the anterior chamber. The ophthalmologist must use all of the available information, age, history of presenting illness, past ocular history, refractive error, and examination findings to distinguish these conditions from true PDS.
Other disorders that have been reported to cause anterior segment pigment dispersion include exfoliation syndrome (XFS), diabetes, herpetic eye disease, iris pigment epithelitis, radiation, trauma, iris pigment epithelial cysts, ciliary body cysts, iris nevus, and melanoma or melanocytoma of the anterior and posterior segment. XFS can be detected by the presence of exfoliation material on the pupillary border or the anterior capsule. Melanomas and melanocytomas can produce unilateral pigment dispersion; however, other signs of intraocular tumor should be present such as iris or ciliary body mass, focal angle closure (atypical for a patient with true PDS), inflammation, or a sentinel scleral vessel. None of these conditions will have the radial, mid-peripheral transillumination iris defects usually seen in PDS.
It is important to remember that the presence of PDS does not preclude other eye conditions in which pigment dispersion may be present. In fact, XFS may be more common in PDS than it is in the general population. The onset of newly elevated IOPs in a patient in his or her sixth decade who has a prior diagnosis of PDS or PG is suggestive of XFS. Patients who may have both PDS and XFS have been described as having the “overlap syndrome.” [20]
Pigmented long anterior zonules (PLAZ), also known as pigmented lens striae, occur when abnormally long and anteriorly inserted zonules are present on the face of the anterior lens capsule, usually bilaterally. This iridozonular apposition creates a special type of pigment dispersion as the zonules rub against the posterior surface of the iris, liberating pigment into the anterior chamber. Krukenberg spindles, densely pigmented trabecular meshwork, and pigmented zonules may be seen on examination. Iris transillumination defects are not typically found in PLAZ, nor is there reverse pupillary block. Unlike PDS, PLAZ is common in black patients, and its incidence increases with hyperopia, age, and female gender [21]. Finally, PLAZ is in some cases associated with a CTRP5 genetic mutation and late-onset macular degeneration [22].
Asymmetric or Unilateral PDS
In general, PDS is bilateral. In all cases of PDS where asymmetric pigment dispersion is present, there is either increased relative pupillary block in the less-involved eye (reducing peripheral iridozonular contact) or greater iridozonular contact in the more-involved eye. Patients will rarely present with heterochromia because of deposition of pigment particles on the iris surface when the involvement is asymmetric. We have observed asymmetric PDS caused by anisometropia, with the less-myopic eye being less involved. Unilateral cataract, anisometropia, trauma, angle recession, and Marfan’s syndrome have been reported in unilateral or highly asymmetric PDS. Two pupillary disorders – Horner’s syndrome and Adie’s pupil – can result in asymmetric PDS, with the involved eye being less pigmented in Horner’s syndrome and more pigmented in Adie’s pupil. In patients in whom no clear cause for asymmetric PDS is found, a more posterior iris insertion and greater iridolenticular contact will be present in the eye with the greater PDS [5]. PDS may resolve partially or completely as a result of cataract extraction, lens subluxation, chronic treatment with miotic therapy, or iridotomy.
Genetics/Inheritance/Epidemiology
A positive family history increases the risk of PDS, supporting the presence of genetic factors in the pathogenesis of PDS and PG. Recessive tyrosinase-related protein 1 (Tyrp1) mutant allele causes iris stromal atrophy in DBA/2J mice with pigment dispersion in the anterior chamber and elevated IOP [23]. However, despite the phenotypic similarity between the pigment dispersion and glaucoma in the DBA/2J mouse and human PDS/PG, DNA sequence variants in the Tyrp1 gene was not associated with inherited human PDS/PG [24]. Single nucleotide polymorphisms (SNPs) in the lysyl oxidase-like 1 (LOXL1) gene have been implicated in the XFS/exfoliative glaucoma [25], which has pigment deposition on the anterior segment structures including trabecular meshwork. However, LOXL1 SNPs were not associated with PDS/PG.
PDS shows autosomal dominant heritability with incomplete penetrance [26]. The most significant risk factors for the development of the phenotypic expressions of PDS are young age, male gender, myopia, European ancestry, and a positive family history. Although men and women are equally affected, men are more likely to develop glaucoma in a ratio of approximately 3:1 [27]. Men are more likely to develop PG from PDS, are diagnosed with PG at an earlier age, and are more likely to require filtering surgery [14].
As a result of obstruction of the intertrabecular meshwork by pigment granules and possible failure or breakdown of normal phagocytic function of trabecular endothelial cells, the IOP elevates in many PDS patients. PDS may be present in up to 2.45 % of the Caucasian population [28]. PDS is rare in blacks and has an estimated prevalence of about 15 cases per 10,000 [29]. In black patients, whose irides may not display classic transillumination defects, infrared pupillography may be helpful. Signs of pigment liberation in blacks (particularly middle-aged black women) may also suggest the PDS-like findings of PLAZ, previously discussed.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


