Persistent Fetal Vasculature
Irene Anteby
David Morrison
Persistent fetal vasculature (PFV) is one of the most common congenital malformations of the human eye. It includes a complex spectrum of clinical manifestations, which develop due to the abnormal persistence of fetal vasculature. The term PFV, coined by Goldberg1 in his 1997 Jackson Memorial Lecture, replaced the more commonly used persistent hyperplastic primary vitreous (PHPV).2 The substitution of the term PFV reflects Goldberg’s more accurate description of anatomic and pathologic features of this disease. In PFV, some, or all, components of the fetal intraocular vasculature remain after birth. This malformation may affect the anterior, retrolental, and/or posterior parts of the infant eye. The extent of the vascular anomaly directly influences both the prognosis for and the therapeutic approach to the PFV eye.
CLINICAL MANIFESTATIONS
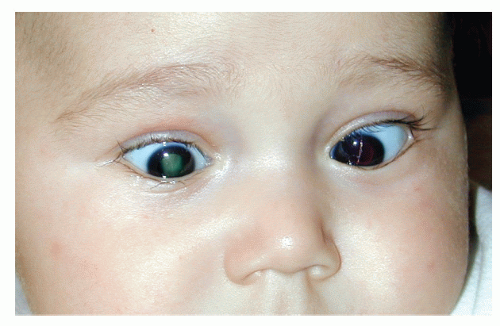
It is important to recognize the range of clinical manifestations in PFV. Knowledge of the embryologic milestones responsible for the development of fetal vasculature is crucial for the understanding of the diversity of symptoms and signs appearing in PFV. During fetal development, transient sets of proliferating blood vessels extend throughout the posterior and anterior poles of the eye. The vessels anastomose freely, creating a rich network by the equator and thus connecting all compartments of the eye. These fetal vessels start to grow during the 1st month of gestation, reach their maximum proliferative activity by the 2nd to 3rd month, and begin to involute at 4 months’ gestation. Normally, these fetal vessels disappear by birth.1,3 In eyes with PFV, the process of fetal vascular regression is arrested. Persistence of some or all fetal vasculature may have profound morphologic consequences. Although individual components of the fetal vasculature often persist in combination with others, any one of the vascular remnants either may predominate in such combinations or may occur alone. Therefore, PFV may cause any of several clinical variants.1
Persistent pupillary membrane. When the tunica vasculosa lentis fails to properly regress, thread-like vessels or pigmented strands may be seen on the lens surface or arising from the iris collarette and attaching to the anterior lens capsule. The pupil may be deformed by these vessels. In rare instances, the entire pupillary axis is obstructed. A thin fibrous sheet may appear, and congenital iris ectropion (entropion uveae) may also occur. Vision may be unaffected or reduced, depending on the extent of pupillary occlusion. The presence of a pupillary membrane may aid in the diagnosis of PFV in an eye with total cataract or whitish retrolental mass.
Iridohyaloid blood vessels. These fetal vessels appear as radial, short, and parallel vessels by the equator of the lens. They constitute a vascular connection between the posterior and the anterior tunica vasculosa lentis. When these vessels do not regress by the second trimester of gestation, they contribute to the appearance of radial superficial vessels in iris stroma. Often, white limbal connective tissue malformation may be seen in the same meridian. When the vessels reach the pupil, they make hairpin loops, inducing a small pupillary notch.
Posterior fibrovascular sheath of the lens. Persistence of the posterior tunica vasculosa lentis may cause the appearance of a fibrovascular mass behind the lens. Reese2 described this as the hallmark of PHPV syndrome. The retrolental membrane may be small or may cover the entire posterior capsule of the lens. It may be associated with a clear lens or cause variable degrees of lenticular opacification. Typically, the retrolental membrane is white or pink in color, differentiating it from the yellow tissue seen in Coats disease or the snow-white tissue typical of calcified retinoblastoma. Formation of a retrolental membrane in PFV is often accompanied by traction on and elongation of the ciliary processes, which may become visible as the pupil is dilated. Although prominent and centrally displaced ciliary processes were once considered pathognomonic for
PHPV, they are also seen in retinopathy of prematurity stage V, Norrie disease, trisomy 13, and congenital subluxated lenses.1
Posterior capsular plaque. The association of posterior capsular plaque and PFV has only recently been described. Mullner-Eidenbock et al.4 first reported that a high proportion of unilateral cataracts had associated findings such as posterior capsular plaque that could represent a subtle form of PFV. The association of posterior capsular plaque and unilateral cataract was later confirmed by the Infant Aphakia Treatment Study (IATS).5 In this multicenter study of primary intraocular lens (IOL) placement versus contact lens use at the time of surgical removal of unilateral congenital cataract, 88% of all children with cataract and 100% of infants with unilateral nuclear cataract had an associated posterior capsular plaque. In the IATS manuscript, plaque was hypothesized to be formed by fetal vessels perforating the lens capsule during lens development. Mullner-Eidenbock et al.4 theorized that these perforating fetal vessels create an abnormally strong adherence of the lens to the posterior capsule. The vessels subsequently resolve, but the lens opacity and posterior capsular plaque remain (Fig. 34.1).
Mittendorf dot. This small white dot on the posterior surface of the lens is typically found 0.5 mm to the nasal side of the center of the posterior pole and designates the point of incomplete regression of the hyaloid artery, where it attaches to the posterior surface of the lens. It is normally found in 0.7% to 2.0% of the population and rarely causes any visual disturbance.3
Persistent hyaloid artery. The fetal hyaloid artery lies within the Cloquet canal and normally loses perfusion around the 7th month of gestation. When this vessel persists, it extends from the optic nerve to the lens. It may be filled with blood but is usually bloodless.

Figure 34.1. Intraoperative photograph of posterior capsular plaque viewed before (A) and after (B) lens removal. Some authors have hypothesized that plaques may be associated with a variant of PFV. (Reprinted from Wilson ME, Trivedi RH, Morrison DG, et al. The Infant Aphakia Treatment Study: evaluation of cataract morphology in eyes with monocular cataracts. J AAPOS 2011;15:421-426, with permission from Elsevier.)
Bergmeister papilla. This term is used to describe a benign remnant of the posterior part of the hyaloid artery that can be seen as an epipapillary vascular tissue. Its effect on vision depends on the presence of other associated optic nerve abnormalities.
Congenital tent-shaped retinal detachment. Congenitally detached retina can result from PFV traction on the retina. It typically has the shape of a traction retinal detachment, and it adheres to the posterior surface of the lens, ciliary body, or both. The detachment may progress, and it has grave visual consequences.
Macular abnormalities. Various dysplastic and hypoplastic abnormalities of the macula may occur in PFV, and these will inevitably affect vision.
Optic nerve abnormalities. Both primary and secondary abnormalities of the optic nerve, including optic disc hypoplasia, may be seen in PFV.
Microphthalmos. Retention of fetal vasculature may be accompanied by an arrest in the growth of the eye globe. Typically, eyes with severe forms of PFV have some degree of microphthalmos. Additional changes include a decreased corneal diameter and distortion of the configuration of globe wall, with colobomatous microphthalmos as a result.
ADJUNCTIVE TOOLS FOR DIAGNOSIS
Despite the wealth of clinical manifestations, diagnosing PFV may sometimes be challenging. Any child with a cataract, unilateral or bilateral, especially when associated with
a microphthalmic globe, should be suspected of having PFV. When PFV is associated with cataract, the differential diagnosis includes diseases causing leukocoria. When clinical signs are nonconclusive, adjunctive imaging may aid in making the correct diagnosis. The most helpful and noninvasive tool is echography. Both posterior segment echography and ultrasound biomicroscopy of the anterior segment6 are valuable. Posterior segment echography typically shows a small globe with a retrolental membrane and a vitreous band extending from the posterior lens capsule to the disc area.7 It can also reveal whether a retinal detachment is present, which may influence the choice of surgical technique used to remove the cataract. High-frequency ultrasonography may demonstrate an anteriorly placed and swollen lens with a resultant shallow anterior chamber, centrally dragged ciliary processes, and thickened anterior vitreous face appearing as a double linear echo near the pars plana or pars plicata.6 In addition, color Doppler imaging of the persistent hyaloid artery may detect blood flow within the stalk (Fig. 34.2). Computerized tomography and magnetic resonance imaging have also been reported as excellent adjunctive devices in the evaluation of PFV.8,9,10 The demonstration of calcifications within the globe is suggestive of retinoblastoma, which is the most important differential diagnosis to rule out in eyes with leukocoria. The rare occurrence of retinoblastoma in an eye with PHPV has been reported.11
a microphthalmic globe, should be suspected of having PFV. When PFV is associated with cataract, the differential diagnosis includes diseases causing leukocoria. When clinical signs are nonconclusive, adjunctive imaging may aid in making the correct diagnosis. The most helpful and noninvasive tool is echography. Both posterior segment echography and ultrasound biomicroscopy of the anterior segment6 are valuable. Posterior segment echography typically shows a small globe with a retrolental membrane and a vitreous band extending from the posterior lens capsule to the disc area.7 It can also reveal whether a retinal detachment is present, which may influence the choice of surgical technique used to remove the cataract. High-frequency ultrasonography may demonstrate an anteriorly placed and swollen lens with a resultant shallow anterior chamber, centrally dragged ciliary processes, and thickened anterior vitreous face appearing as a double linear echo near the pars plana or pars plicata.6 In addition, color Doppler imaging of the persistent hyaloid artery may detect blood flow within the stalk (Fig. 34.2). Computerized tomography and magnetic resonance imaging have also been reported as excellent adjunctive devices in the evaluation of PFV.8,9,10 The demonstration of calcifications within the globe is suggestive of retinoblastoma, which is the most important differential diagnosis to rule out in eyes with leukocoria. The rare occurrence of retinoblastoma in an eye with PHPV has been reported.11
PFV AND ASSOCIATED ANOMALIES
Although PFV mostly appears as a single anomaly, sometimes it may be associated with other ocular abnormalities such as Peters anomaly,12 Rieger anomaly,13 and morning glory syndrome.14,15 Only 5% to 10% of children with PFV have binocular involvement. Bilaterality represents a more widespread degree of abnormal embryologic development. Associated systemic anomalies may occur, especially neurologic abnormalities.16 Haddad et al.17 reported systemic abnormalities including cleft palate and lip, polydactyly, and microcephaly in association with bilateral PHPV. Goldberg reported on the association of PFV with trisomy 13.1 A few pedigrees with familial PFV have been described,18,19 suggesting the possibility of an autosomal recessive20 or autosomal dominant21 inheritance pattern in selected cases. In animal models, the presence of PFV has been associated with Arf tumor suppressor gene deficiency,22 angiopoietin-2 deficiency,23 abnormalities of macrophage-induced programmed cell death,24 and abnormalities of astrocyte cell migration.25
 Get Clinical Tree app for offline access 
|


