21 PEDIATRICS
Amblyopia
Suzanne M. Wickum
ICD-9: 368.01—AMBLYOPIA, STRABISMIC
ICD-9: 368.02—AMBLYOPIA, DEPRIVATION
ICD-9: 368.03—AMBLYOPIA, REFRACTIVE
 THE DISEASE
THE DISEASE
Pathophysiology
Amblyopia is a unilateral or bilateral decrease in visual acuity, uncorrectable by optical means, without detectable anatomic damage in the eye or visual pathway. When form vision deprivation and abnormal binocular interaction occur during the critical or sensitive periods, amblyopia may develop.
Etiology
Amblyopia is a diagnosis of exclusion. In addition to ruling out ocular pathology, an amblyogenic factor must be present. There are four main etiologic categories of amblyopia.
Strabismic Amblyopia
- Abnormal binocular interaction secondary to constant or intermittent strabismus.
- Moderate to deep amblyopia is associated with constant, unilateral strabismus.
- Shallow amblyopia is possible with frequent intermittent, unilateral strabismus.
Refractive Amblyopia
- Isoametropic
1. Bilateral visual disruption because of significant uncorrected ametropia.
2. Bilateral hyperopia greater than 2 to 4 D, myopia greater than 6 to 8 D, and astigmatism greater than 1.50 to 2.50 D can cause isoametropic amblyopia.
- Anisometropic
1. Unilateral visual disruption because of significant uncorrected anisometropia.
2. The greater the amount of anisometropia, the deeper the amblyopia.
3. Uncorrected hyperopic anisometropia greater than 3.50 D and myopic anisometropia greater than 6.50 D lead to a 100% incidence of amblyopia.
4. Uncorrected hyperopic anisometropia of 2.00 to 3.50 D and myopic anisometropia of 5.00 to 6.50 D lead to a 50% incidence of amblyopia.
- Meridional
1. Uncorrected astigmatism leads to meridional visual deprivation.
2. Uncorrected astigmatism of 1.50 to 2.00 D or more can be amblyogenic.
Combined Anisometropic-Strabismic Amblyopia
- Both anisometropia and strabismus are present.
- The strabismic eye is generally the more ametropic eye.
Deprivation Amblyopia
- Unilateral or bilateral visual deprivation because of the obstruction of the visual axis by ptosis, corneal opacity, cataracts, or other media opacities. May also be iatrogenic.
- Often causes very deep amblyopia.
Clinical Symptoms
- Decreased vision in one or both eyes.
Clinical Signs
- Reduced best-corrected visual acuity in one or both eyes in the absence of ocular disease and in the presence of an amblyogenic factor.
- Abnormal contour interaction (crowding phenomenon) in the amblyopic eye.
- Eccentric fixation (usually associated with strabismic amblyopia).
- Decreased contrast sensitivity in the amblyopic eye.
- Binocular suppression of the amblyopic eye.
- Reduced accommodative response in the amblyopic eye.
- Mild afferent pupillary defects have also been reported in some severely amblyopic eyes.
Demographics
Amblyopia occurs in 2% to 4% of the general population. Amblyopia is the most common visual disability in children, with approximately 120,000 children diagnosed per year in the United States. Amblyopia is the leading cause of monocular vision loss in the 20- to 70-year age group.
Significant History
- A history of “lazy eye,” misaligned eyes, uncorrected ametropia, extraocular muscle surgery, occlusion therapy, or other vision therapy may be elicited.
Ancillary Tests
A thorough eye examination is indicated with particular attention to best-corrected visual acuity, binocularity, cycloplegic retinoscopy/refraction, and ocular health. Visuoscopy is a useful ancillary test to determine the presence or absence of eccentric fixation.
The Treatment
Strabismic Amblyopia
- Optical correction of ametropia based on cycloplegic retinoscopy/refraction.
- Part-time, direct, full occlusion for 2 to 6 h/d for mild to moderate amblyopia, with the possible need for increased hours for deep amblyopia.
- Full-time occlusion may be necessary when trying to break down and/or prevent binocular sensory anomalies (suppression, anomalous correspondence); however, care must be taken to prevent occlusion amblyopia. For patients 5 years and under, occlude the preferred eye the number of days equal to the child’s age, and then occlude the amblyopic eye for 1 day.
- Penalization with 1% atropine may be used instead of traditional occlusion for mild to moderate amblyopia.
- Active monocular visual activities for 30 to 60 min/d while occluded.
- Follow up every 4 to 6 weeks.
- Manage the strabismus once the amblyopia treatment is completed or abandoned.
- Active monocular visual activities for 30 to 60 min/d while occluded.
Isoametropic Amblyopia
- Optical correction of ametropia based on cycloplegic retinoscopy/refraction.
- Follow up in approximately 4 weeks.
- If necessary, make any changes to the prescription.
- Follow every 4 to 6 months to monitor acuity improvement.
- Patching or penalization is necessary only in cases of asymmetric aided acuity.
Anisometropic Amblyopia
- Optical correction of ametropia based on cycloplegic retinoscopy/refraction.
- Part-time, direct, full occlusion for 2 to 6 h/d for mild to moderate amblyopia, with the possible need for increased hours for deep amblyopia.
- Penalization with 1% atropine may be used instead of traditional occlusion for mild to moderate amblyopia.
- Active monocular visual activities for 30 to 60 min/d while occluded. Once visual acuity in the amblyopic eye reaches 20/40–50, consider the addition of binocular vision therapy to break down remaining suppression.
- Follow up every 4 to 6 weeks.
Deprivation Amblyopia
- Remove the obstruction of the visual axis promptly, ideally within 2 months of onset.
- Correct any significant ametropia or aphakia.
- Part-time, direct, full occlusion for 2 to 6 h/d for mild to moderate amblyopia, with the possible need for increased hours for deep amblyopia.
- Penalization with 1% atropine may be used instead of traditional occlusion for mild to moderate amblyopia.
- Active monocular visual activities for 30 to 60 min/d while occluded.
- Follow up every 4 to 6 weeks.
Esodeviations
Suzanne M. Wickum
ICD-9: 378.00—ESOTROPIA, UNSPECIFIED
ICD-9: 378.01—ESOTROPIA, MONOCULAR
ICD-9: 378.02—ESOTROPIA, MONOCULAR WITH “A” PATTERN
ICD-9: 378.03—ESOTROPIA, MONOCULAR WITH “V” PATTERN
ICD-9: 378.04—ESOTROPIA, MONOCULAR WITH OTHER NONCOMITANCY
ICD-9: 378.05—ESOTROPIA, ALTERNATING
ICD-9: 378.06—ESOTROPIA, ALTERNATING WITH “A” PATTERN
ICD-9: 378.07—ESOTROPIA, ALTERNATING WITH “V” PATTERN
ICD-9: 378.08—ESOTROPIA, ALTERNATING WITH OTHER NONCOMITANCY
ICD-9: 378.21—ESOTROPIA, INTERMITTENT, UNILATERAL
ICD-9: 378.22—ESOTROPIA, INTERMITTENT, ALTERNATING
ICD-9: 378.35—ESOTROPIA, WITH ACCOMMODATIVE COMPONENT
 THE DISEASE
THE DISEASE
Pathophysiology
Esodeviations present with an intermittent or constant inward eye turn resulting in convergent visual axes.
Etiology
Innervational, accommodative, genetic, and environmental factors play a role in the development of esotropia.
The Patient
Clinical Symptoms
- Patients may report diplopia, asthenopia, and/or blurred vision.
Esotropia Classification and Characteristics
Infantile Esotropia
- Clinical characteristics: Onset occurs between birth and 6 months of age. Esotropia is constant, comitant (A or V patterns may be present), moderate to large magnitude (30 to 70 prism diopters), and cross-fixation is common. The accommodative-convergence accommodation (AC/A) ratio is usually normal. Ametropia is skewed toward low hyperopia. Sensory adaptations are present in 35% to 72% of cases. Dissociated vertical deviation is present in 50% to 75% of cases. Overaction of the inferior oblique muscle is present in 68% of cases. Latent or manifest-latent nystagmus is found in 25% to 52% of cases.
Accommodative Esotropia
Refractive Accommodative Esotropia
- Clinical characteristics: Onset is usually between 1 and 8 years of age with an average of 2.5 years. Deviation is typically comitant (A or V patterns may be present), moderate (20 to 40 prism diopters), variable, and intermittent with a gradual increase in frequency and duration over time. If the deviation becomes constant, sensory adaptations may develop. The AC/A ratio is normal. The etiology is uncorrected hyperopia and insufficient fusional divergence. Ametropia is usually between +2 and +6 D with an average of +4.75 D.
Nonrefractive Accommodative Esotropia
- Clinical characteristics: The characteristics are the same as refractive accommodative esotropia with a few exceptions. The AC/A ratio in this case is high, and thus the near angle is larger than distance. The average ametropia is +2.25 D, but any refractive error can be found. The etiology in this case is the high AC/A ratio; therefore, esotropia typically responds to plus lenses at near.
Partially Accommodative Esotropia
- Clinical characteristics: The characteristics are the same as refractive accommodative esotropia with a few exceptions. Esotropia is constant; therefore, sensory adaptations are likely. The AC/A ratio is normal or high. Typically, there is moderate to high hyperopia present. Esotropia magnitude decreases, but is not eliminated, with the hyperopic correction.
Nonaccommodative Acquired Esotropia
- Clinical characteristics: Onset occurs after 6 months of age. Esotropia is typically constant, comitant (A or V patterns may be present), and of moderate to large magnitude (20 to 70 prism diopters), and sensory adaptations may occur. Often, some amount of hyperopia is present. Causes include idiopathic, decompensated esophoria; disruption of fusion by occlusion; physical/emotional stress; and rarely CNS pathology.
Basic Esotropia
- This is the most common subcategory. The AC/A ratio is normal. The refractive error is usually insignificant.
Divergence Insufficiency Esotropia
- The AC/A ratio is low, and thus the distance magnitude is greater than near. Divergence paralysis, which originates from a midbrain lesion, must be ruled out. Divergence paralysis initially presents as a noncomitant esotropia that shows spread of comitancy.
Acute Esotropia
- Diplopia is likely. Some patients may close or wink one eye to alleviate the diplopia.
Sensory Esotropia
- Onset may occur at any age. Deviation is constant, comitant, and unilateral, and the magnitude is often variable. The strabismus is secondary to significantly reduced visual acuity, resulting in a sensory obstacle to fusion. Visually depriving factors may include uncorrected anisometropia, ptosis, corneal opacities, cataracts, optic nerve lesions, or retinal lesions. Sensory esotropia is as frequent as sensory exotropia in children under 6 years of age; however, sensory exotropia predominates in older children and adults.
Microtropia
Clinical Characteristics
- Primary microtropia is likely present since birth, with no history of a larger angle of strabismus. Secondary microtropia is often the result of vision therapy or surgery for a larger deviation. Other causes may include aniseikonia, anisometropia, uncorrected vertical deviation, and foveal lesions. Deviation is usually constant and comitant, and the magnitude is between 1 and 10 prism diopters. Sensory adaptations are common and include anomalous correspondence, mild amblyopia, central suppression, and eccentric fixation. These patients often have some peripheral fusion with anomalous vergence ranges and some local stereopsis but no global stereopsis.
Incomitant Esotropia
Abducens Nerve Palsy (Sixth Cranial Nerve Palsy)
- A noncomitant esotropia with greatest magnitude in the affected muscle action field. The distance magnitude may be larger than near. An abduction deficit is present, with no restriction on forced duction testing. Patients typically report diplopia and may adopt a face turn toward the paretic eye to obtain fusion. (See Chapter 19, “Neuro-ophthalmic Disease.”)
Duane Syndrome and Mobius Syndrome
- See Strabismus Syndromes later in this chapter.
Demographics
The prevalence of strabismus in the general population is between 2% and 6%. Esotropia occurs three times as often as exotropia. While most cases of esotropia have an accommodative component, 28% to 54% of esotropias are infantile.
Significant History
- Age and nature of onset
- Frequency of deviation
- Changes in the size or frequency of the deviation
- Associated symptoms (including diplopia)
- History of injury or illness
- History of neurologic, systemic, or developmental disorders
- History of ocular disease or reduced visual acuity
- History and outcome of prior treatment (occlusion, vision therapy, prism, surgery)
- Positive family history of strabismus
Ancillary Tests
A thorough ocular examination is indicated in all strabismic patients. Careful refractive error and visual acuity measurements should be performed to evaluate for amblyopia. Visuoscopy can be utilized to detect eccentric fixation. Measurement of the esotropia including frequency, laterality, magnitude, and comitancy is essential. Evaluation of ocular motility is necessary to rule out abducens palsy. In cases of intermittent esotropia, base-in (compensating) vergence ranges should be measured and sensory-motor fusion should be assessed (Worth-dot/stereopsis). Subjective assessment of the deviation is performed with tests such as Maddox rod and Bagolini lenses. The subjective angle of deviation is compared to the objective angle of deviation to determine the presence/absence of anomalous correspondence. Cycloplegic retinoscopy/refraction is recommended. Lastly, a thorough ocular health examination is necessary to rule out causes for sensory deprivation.
The Treatment
- Treatment of infantile esotropia starts with correction of any significant refractive error. When amblyopia is present, it should be treated prior to surgical intervention. Once the patient can freely alternate fixation, strabismus surgery should be offered. Ideally, surgery should be performed by 6 to 12 months of age for the best chance of establishing some form of binocularity.
- Treatment of refractive/accommodative acquired esotropia includes prescribing the full hyperopic correction. Consider giving an add for cases with a high AC/A ratio. Treat amblyopia and sensory-motor fusion as needed. If the esotropia is only partially accommodative, consider adding base-out prism when the residual esotropia is less than 15 to 20 prism diopters and surgical intervention if greater than 15 to 20 prism diopters.
- Once the etiology of nonaccommodative acquired esotropia has been determined, treatment can begin. If amblyopia is present, it should be treated. For esotropia less than 15 to 20 prism diopters, base-out prism and/or vision therapy should be utilized. For deviations greater than 15 to 20 prism diopters, surgery should be considered.
- Microtropia treatment is often confined to best optical correction and treatment of amblyopia. When anomalous correspondence is present, binocular therapy is rarely undertaken; however, when normal correspondence is present, base-out prism and/or vision therapy can be utilized to obtain sensory-motor fusion.
- In the case of sensory esotropia, the underlying cause must be addressed first. Any significant ametropia should be corrected, and all patients should wear protective glasses full-time. If superimposed amblyopia is suspected, treatment should be implemented. If improved ocular alignment is desired, strabismus surgery may be performed.
- Microtropia treatment is often confined to best optical correction and treatment of amblyopia. When anomalous correspondence is present, binocular therapy is rarely undertaken; however, when normal correspondence is present, base-out prism and/or vision therapy can be utilized to obtain sensory-motor fusion.
Exodeviations
Suzanne M. Wickum
ICD-9: 378.10—EXOTROPIA, UNSPECIFIED
ICD-9: 378.11—EXOTROPIA, MONOCULAR
ICD-9: 378.12—EXOTROPIA, MONOCULAR WITH ‘A’ PATTERN
ICD-9: 378.13—EXOTROPIA, MONOCULAR WITH ‘V’ PATTERN
ICD-9: 378.14—EXOTROPIA, MONOCULAR WITH OTHER NONCOMITANCY
ICD-9: 378.15—EXOTROPIA, ALTERNATING
ICD-9: 378.16—EXOTROPIA, ALTERNATING WITH ‘A’ PATTERN
ICD-9: 378.17—EXOTROPIA, ALTERNATING WITH ‘V’ PATTERN
ICD-9: 378.18—EXOTROPIA, ALTERNATING WITH OTHER NONCOMITANCY
ICD-9: 378.23—EXOTROPIA, INTERMITTENT, UNILATERAL
ICD-9: 378.24—EXOTROPIA, INTERMITTENT, ALTERNATING
 THE DISEASE
THE DISEASE
Pathophysiology
Exodeviations present as an intermittent or constant outward eye turn resulting in divergent visual axes. Deviation often begins as an exophoria and over time becomes an intermittent exotropia. When left untreated, an intermittent exotropia may progress to a constant exotropia.
Etiology
Innervational, mechanical/anatomic, and multifactorial genetic inheritance patterns have all been implicated in the development of exotropia.
The Patient
Clinical Symptoms
- Patients may complain of asthenopia, squinting, photophobia, unilateral eye closure, diplopia, and/or blurred vision.
Exotropia Classification and Characteristics
Primary Comitant Exotropia
Clinical Characteristics
- Onset is usually between 6 months and 8 years of age. Exotropia is typically intermittent (80%) and comitant, although A or V patterns are found with overaction of the superior or inferior oblique muscles. Deviation magnitude ranges from 20 to 70 prism diopters. Positive fusional vergence is insufficient. Sensory adaptations are minimal. Fatigue, illness, daydreaming, alcohol/sedatives, or going from dim to bright light (“dazzle effect”) may decrease control of the exotropia.
- Subcategories of primary comitant exotropia are as follows (based on Duane’s categories):
1. Basic exotropia: Distance and near deviation magnitude are similar, yielding a normal AC/A ratio. Exotropia is manifest more often at distance than at near.
3. True divergence excess exotropia: Distance deviation magnitude is greater than near, yielding a high AC/A ratio. Exotropia is more frequent at distance.
4. Pseudo-divergence excess exotropia: Initial measurements look like divergence excess exotropia with the strabismus magnitude greatest at distance. These patients have increased tonic fusional convergence that is not broken down with a near cover test. With prolonged occlusion, or with +3.00 D overcorrection, the magnitude of the near deviation is found to be similar to the distance deviation, resulting in a basic exotropia. Pseudo-divergence excess is more common than true divergence excess.
Infantile Exotropia
Clinical Characteristics
- A rare type of exotropia occurring in approximately 1/30,000 births. Onset occurs between birth and 6 months of age. Deviation is constant, moderate to large (30 to 90 prism diopters), and comitant with possible A or V patterns if superior or inferior oblique overactions are present. Sensory adaptations are likely. Infantile exotropia may occur in otherwise healthy infants but is typically associated with other ocular disorders, neurologic disease, craniofacial syndromes, and genetic syndromes.
Sensory Exotropia
Clinical Characteristics
- Onset may occur at any age. Deviation is constant, unilateral, and comitant, and the magnitude is often variable. The strabismus is secondary to significantly reduced visual acuity, resulting in a sensory obstacle to fusion. Visually depriving factors may include uncorrected anisometropia, ptosis, corneal opacities, cataracts, optic nerve lesions, or retinal lesions. Sensory exotropia is as frequent as sensory esotropia in children under 6 years of age; however, sensory exotropia predominates in older children and adults.
Oculomotor Nerve Palsy (Third Cranial Nerve Palsy)
Clinical Characteristics
- A large magnitude, noncomitant exotropia with hypotropia. Accompanied by limitation of elevation, depression, and adduction in the affected eye. A complete palsy also presents with ptosis, loss of accommodation, and a dilated pupil. These findings vary in the case of an incomplete palsy. (See Chapter 19, “Neuro-ophthalmic Disease.”)
Demographics
The prevalence of strabismus in the general population is between 2% and 6%. Exodeviations occur less often than esodeviations in a 1:3 ratio. In addition, exodeviations occur more often in females than males. Approximately one third of the exodeviations are evident by 2 years of age.
Significant History
- Age and nature of onset
- Frequency of deviation
- Changes in the size or frequency of the deviation
- Associated symptoms (including diplopia)
- History of injury or illness
- History of neurologic, systemic, or developmental disorders
- History of ocular disease or reduced visual acuity
- History and outcome of prior treatment (occlusion, vision therapy, prism, surgery)
- Positive family history of strabismus
Ancillary Tests
A thorough ocular examination is indicated in all strabismic patients. Careful refractive error and visual acuity measurements should be performed to evaluate for amblyopia. Visuoscopy can be utilized to measure eccentric fixation. Measurement of the exotropia including frequency, laterality, magnitude, and comitancy is essential. Evaluation of ocular motility and near point of convergence is necessary. In cases of intermittent exotropia, base-out (compensating) vergence ranges should be measured, and sensory-motor fusion should be assessed (Worth-dot/stereopsis). Subjective assessment of the deviation is performed with tests such as Maddox rod and Bagolini lenses. The subjective angle of deviation is then compared to the objective angle of deviation to determine the presence/absence of anomalous correspondence. Cycloplegic retinoscopy/refraction is recommended in all strabismic patients. A thorough ocular health examination is necessary to rule out causes of sensory deprivation.
The Treatment
- For all classifications of exotropia, the initial treatment should include correcting significant refractive error and treating amblyopia.
- Minus lenses may be used full-time as a passive therapy for young children or may be utilized during vision therapy sessions for older children and adults.
- Relieving or correcting prism (base-in) may be used as an individual treatment or may be combined with vision therapy.
- Vision therapy can be utilized in some cases of exotropia. The therapy begins by increasing the patient’s gross convergence ability, thus normalizing the near point of convergence. Next, sensory-motor fusion is trained with the goal of expanding the patient’s positive fusional vergence range so as to compensate for the exotropia. Therapy techniques that enhance diplopia awareness and break suppression are recommended.
- Surgery is utilized in moderate- to large-angle exotropia. In cases of intermittent exotropia, many surgeons recommend that surgery be delayed until 4 years of age, unless the frequency of the exotropia is increasing. In the case of constant exotropia, surgery should be performed much earlier.
- Utilizing vision therapy before and/or after surgery improves the chances for functional binocular vision.
- For cases of sensory exotropia, the treatment is different. First, the underlying cause of the reduced acuity must be addressed. Any significant ametropia should be corrected, and all patients should be given protective glasses for full-time wear. If superimposed amblyopia is suspected, treatment should be implemented. If improved ocular alignment is desired, strabismus surgery may be performed.
Strabismus Syndromes
Suzanne M. Wickum
ICD-9: 378.71—DUANE RETRACTION SYNDROME
ICD-9: 378.61—BROWN’S SYNDROME
ICD-9: 352.6—MOBIUS SYNDROME
ICD-9: 378.62—EYE MUSCLE FIBROSIS
DUANE RETRACTION SYNDROME (DRS)
 THE DISEASE
THE DISEASE
Pathophysiology
Duane retraction syndrome is a congenital ocular motility disorder characterized by globe retraction and narrowing of the palpebral fissure in attempted adduction, frequent abduction deficiency, variable adduction deficiency, and often upshoots or downshoots upon attempted adduction.
Etiology
Although there have been many theories regarding the etiology of DRS, the most widely accepted theory involves innervational and central nervous system anomalies. It has been found that the abducens nucleus and nerve are absent or hypoplastic, resulting in limited or absent abduction. Additionally, there is anomalous innervation of the lateral rectus muscle by branches of the oculomotor nerve.
Most cases of DRS are sporadic; however, familial cases have been reported to occur in 5% to 23% of patients. The inheritance pattern is autosomal dominant with incomplete penetrance and variable expressivity. Since DRS is associated with a 10 to 20 times increase in frequency of other congenital ocular and nonocular anomalies, there is speculation that a teratogenic event may take place somewhere between the 4th and 10th week of gestation in sporadic cases.
The Patient
Clinical Symptoms
Because of the young age at diagnosis, the patient will not be symptomatic; however, the parents may report that the child has an eye turn, abnormal face turn, and/or abnormal eye movements.
Clinical Signs
DRS is divided into three categories. Each type has the common characteristics of globe retraction and narrowing of the palpebral fissure on adduction, as well as upshoots or downshoots on attempted adduction.
DRS type I characteristics: Esotropia or heterophoria are common in primary gaze, but exotropia is possible. There is marked limitation of abduction with no/minimal limitation of adduction. The child may adopt a face turn in the direction of the abduction deficit in order to maintain binocularity.
DRS type II characteristics: Exotropia is frequently observed in primary gaze. There is marked limitation of adduction with no/minimal limitation of abduction. The child may adopt a face turn in the direction of the adduction deficit in order to maintain binocularity.
DRS type III characteristics: Exotropia is frequently observed in primary gaze. There is combined limitation of abduction and adduction. Face turns are variable.
Demographics
Duane retraction syndrome is the most common of all congenital oculomotor anomalies and accounts for 1% to 4% of strabismus cases. While there is no racial predilection, there is a slight preponderance for females (55% to 60% of cases). The left eye is affected in approximately 60% of cases, the right eye in 22%, and bilateral in 18%. DRS type I is the most common (78%), followed by DRS type III (15%) and DRS type II (7%).
Significant History
- Age of onset
- Positive family history of ocular motility restriction/strabismus
- Presence of diplopia (DRS patients rarely report diplopia)
Ancillary Tests
Evaluation of ocular misalignment, abnormal face turn, range of extraocular motility, globe retraction, palpebral fissure narrowing, and observation of upshoots and downshoots are important. In difficult cases, forced ductions are useful in differentiating between long-standing muscle problems with secondary fibrosis versus recently acquired cranial nerve palsies.
The Treatment
Surgery is utilized to improve unacceptable face turns, correct significant ocular misalignment in primary gaze, reduce severe globe retraction, and improve the appearance of upshoots and downshoots. It must be stressed that surgery may improve, but will not eliminate, the abnormal eye movements. For patients who are not good surgical candidates but who have asthenopia, vision therapy and/or prism can be beneficial.
BROWN’S SYNDROME (SUPERIOR OBLIQUE TENDON SHEATH SYNDROME)
 THE DISEASE
THE DISEASE
Pathophysiology
Brown’s syndrome is typically a congenital ocular motility disorder, although acquired forms are possible. The syndrome is primarily characterized by deficient elevation in adduction.
Etiology
In congenital Brown’s syndrome, the motility deficiency is constant and not likely to resolve. The motility deficiency is a result of a short or inelastic superior oblique tendon or because of a limitation of the tendon to slide through the pulley-like trochlea. Most cases of congenital Brown’s syndrome are sporadic; however, familial cases have been reported. The inheritance pattern is autosomal dominant with incomplete penetrance and variable expressivity.
In acquired Brown’s syndrome, the motility deficiency may be constant or intermittent and may resolve over time. This syndrome may be caused by surgical procedures including strabismus surgery, sinus surgery, blepharoplasty, and scleral buckling. Brown’s syndrome is also associated with inflammatory disorders such as rheumatoid arthritis, sinusitis, and metastasis in the area of the trochlea. Trauma may also give rise to Brown’s syndrome.
The Patient
Clinical Symptoms
Patients may complain of pain or tenderness in between their eyes (in the area of the trochlea), especially during up gaze movements. Some patients may also report an audible “clicking” noise as they look up. Additionally, patients may complain of diplopia, particularly in up gaze.
Clinical Signs
There is limitation or absence of elevation in adduction with normal/near normal elevation in midline and abduction. Hypotropia of the involved eye may be found in primary gaze. Divergence occurs in up gaze, causing a V-pattern deviation. Downshoots on adduction are possible. There is minimal or no superior oblique overaction of the ipsilateral eye. Patients may adopt a compensatory chin-up head posture to preserve binocularity and/or eliminate diplopia.
Demographics
Brown’s syndrome occurs in less than 1% of strabismus patients. It has been estimated to occur in 1/20,000 live births. This condition does not show any gender or racial predilection. Brown’s syndrome is found unilaterally in 90% of cases and bilaterally in 10% of cases.
Significant History
- Age of onset
- Positive family history of ocular motility restriction/strabismus
- Presence of diplopia
- Recent history of strabismus surgery, sinus surgery, retinal surgery, or blepharoplasty
- History of inflammatory conditions such as rheumatoid arthritis or sinusitis
- History of trauma
- History of inflammatory conditions such as rheumatoid arthritis or sinusitis
Ancillary Tests
Evaluation of ocular misalignment, abnormal head posture, range of extraocular motility, and observation of downshoots are important. Additionally, forced ductions are useful in confirming the restrictive nature of this syndrome.
The Treatment
In many cases of Brown’s syndrome, observation and patient/parent education are all that are necessary. This is particularly true in mild cases with preserved binocularity in primary gaze and without an anomalous head posture. Indications for surgical intervention include hypotropia present in primary gaze, significant anomalous head posture, and/or unacceptable downshoots of the involved eye. In inflammatory cases, steroids administered orally or locally are useful.
MOBIUS SYNDROME
 THE DISEASE
THE DISEASE
Pathophysiology
Mobius’ syndrome consists of congenital facial diplegia and bilateral abducens nerve palsies. Systemic abnormalities and other cranial nerve palsies may also be present.
Etiology
The exact pathogenesis of Mobius syndrome is uncertain; however, the timing of the syndrome seems to be well defined. A pathologic insult takes place during the first trimester, typically between the fourth to sixth week of gestation, when the cranial nerve nuclei are undergoing rapid development. Trauma, illness, and maternal ingestion of certain medications have been associated with this syndrome. In more recent years, a vascular etiology of Mobius syndrome has been implicated. The vascular insult leads to hypoxia and ischemia of the cranial nerve nuclei.
While the majority of Mobius syndrome cases are sporadic, there have been case reports of pedigrees demonstrating autosomal dominant, autosomal recessive, and X-linked inheritance.
The Patient
Clinical Symptoms
Symptoms present in infancy and include difficulty sucking, excessive drooling, lack of facial expression, and lagophthalmos. Parents may also report crossed eyes and tracking problems.
Clinical Signs
Common ocular signs include esotropia, bilateral abduction deficits, lagophthalmos, and exposure keratitis. Additional ocular or orbital signs have been noted, including hypertelorism, epicanthal folds, small palpebral fissures, ptosis, and entropion. Abnormalities of the extremities, swallowing and speech difficulties, craniofacial abnormalities, defective brachial musculature, tongue hypoplasia, and mental retardation have all been reported as well.
Demographics
Mobius syndrome is very rare. No racial or gender predilections have been reported.
Significant History
- Trouble with sucking/feeding
- Eyes partially open when sleeping
- Presence of photophobia
- Problems with facial expression
A complete eye examination is necessary, with special attention to eye alignment, extraocular motility, lagophthalmos, and exposure keratitis. When esotropia is present, one must also evaluate the patient for amblyopia. Because of the frequently associated systemic anomalies, the patient should also have a complete physical examination with a pediatrician.
The Treatment
Ocular management includes correcting significant refractive error, providing therapy for amblyopia, considering strabismus surgery when significant esotropia is present, and treating keratitis associated with lagophthalmos. A pediatrician should manage any systemic anomalies.
CONGENITAL FIBROSIS OF THE EXTRAOCULAR MUSCLES (CFEOM)
 THE DISEASE
THE DISEASE
Pathophysiology
CFEOM is a rare disorder characterized by congenital restrictive ophthalmoplegia.
Etiology
CFEOM is typically a familial disorder inherited in an autosomal dominant pattern with significant variability in expression; however, cases of autosomal recessive inheritance as well as sporadic cases have been reported. Original reports describe CFEOM as having a myogenic origin. More recent reports indicate a primary neurogenic etiology that leads to subsequent degeneration of muscle fibers.
The Patient
Clinical Symptoms
Because of the young age at diagnosis, the patient will not be symptomatic; however, parents will report that the child has abnormal eye movements, abnormal eye alignment, ptosis, and/or an abnormal head posture.
Clinical Signs
Traditionally, this syndrome has been divided into five categories based on slight clinical differences. The general fibrosis syndrome is characterized by bilateral restriction of all of the extraocular muscles. The inferior rectus shows greatest involvement, thus causing the eyes to be fixed approximately 20 to 30 degrees below midline. As a result of the bilateral hypotropic eye position combined with bilateral ptosis, the patient will adopt a chin-up head position. Congenital fibrosis of the inferior rectus with blepharoptosis is a variant of CFEOM in which the inferior rectus is affected with little to no involvement of the other extraocular muscles. In cases of strabismus fixus, the eyes are fixed in either an esotropic or exotropic position with severe limitation of horizontal eye movements. The vertical eye movements are usually intact. In contrast, cases of vertical retraction syndrome demonstrate severely impaired vertical eye movements with relatively intact horizontal movements. Lastly, a unilateral fixed globe characterizes congenital unilateral fibrosis.
More recently, a revised classification has been proposed based on the identification of three CFEOM genetic loci. Patients with CFEOM1, mapped to chromosome 12, have the features of general fibrosis syndrome as listed above. Cases with CFEOM2, mapped to chromosome 11, present with exotropic strabismus fixus. Patients with CFEOM3, mapped to chromosome 16, present with variable expression of ptosis and restrictive ophthalmoplegia.
Demographics
CFEOM is a rare disorder. The prevalence of CFEOM is relatively unknown, although one report from the United Kingdom estimates this condition to occur in 1 of 230,000 people. There are no reports of racial or gender predilection.
Significant History
- Age of onset
- Positive family history of similar ocular motility restrictions and ptosis
Ancillary Tests
Forced duction testing is useful in confirming the restrictive nature of this condition. Orbital imaging such as computed tomography or magnetic resonance imaging may be of benefit. Imaging studies may show extraocular muscle atrophy, abnormal insertion, or absence.
The Treatment
Treatment for CFEOM is primarily surgical; however, proper refractive correction and treatment of amblyopia should be done prior to surgical intervention.
Leukocoria
Suzanne M. Wickum
ICD-9: 360.44
 THE DISEASE
THE DISEASE
Pathophysiology
Leukocoria literally means white pupil. Leukocoria itself is not a specific diagnosis, but rather a clinical finding suggestive of intraocular pathology. When an ocular condition disrupts light from striking the retina, a whitish reflex, rather than the usual red, will be evident in the pupil.
Etiology
- The most common cause of leukocoria in children is cataracts. These lens opacities may be unilateral or bilateral. With congenital cataracts, one third are inherited, one third are associated with systemic disorders or syndromes, and one third are idiopathic. Diagnosis is typically made at birth or soon after. Acquired cataracts in children have a variety of causes including trauma, uveitis, metabolic disorders, and drug induced.
- The most common primary malignant intraocular tumor in children is retinoblastoma. Leukocoria is the most common presenting sign. This tumor appears as a yellowish-white, nodular mass with overlying and/or intralesional vascularization. Strabismus, glaucoma, retinal detachment (RD), vitreous seeding, vitreal hemorrhage, pseudohypopyon, hyphema, proptosis, and even orbital cellulitis may occur. Tumors may be unilateral or bilateral. The average age at diagnosis is between 12 and 21 months.
- Coats disease is a sporadic retinal telangiectasis that may lead to exudative RDs giving rise to the appearance of leukocoria. It is predominantly found in male (80%) and is typically unilateral (90%). Onset is usually between 8 and 10 years of age, although presentation in infancy has been reported. Advanced cases of Coats’ disease can be difficult to differentiate from retinoblastoma by clinical examination alone.
- Persistent hyperplastic primary vitreous (PHPV), also known as persistent fetal vasculature, is a congenital abnormality caused by failure of the primary vitreous to regress. This condition is typically unilateral (90%) and sporadic. PHPV presents as a white fibrovascular membrane or mass adherent to the back of the lens. Leukocoria can result from the retrolental opacity or from a secondary cataract. Other features of PHPV include microphthalmia, microcornea, shallow anterior chamber, dilated iris vessels, elongated ciliary processes, RD, and secondary angle-closure glaucoma.
- Retinopathy of prematurity (ROP) is a vasoproliferative retinopathy that occurs in premature and low–birth weight infants. Infants at risk include those born at less than 32 weeks of gestation and/or weighing less than 1,500 to 2,000 g. Diagnosis is typically made at 4 to 6 weeks of age. Leukocoria is because of RD found in the advanced stages of ROP.
- Toxocariasis is caused by infection with the larvae of Toxocara canis. Toxocara is unilateral and found more frequently in males than females. Frequently, there is a history of contact with puppies, eating dirt, or playing in a sandbox. The mean age of presentation is 7 years. Characteristics of toxocariasis include a white retinal granuloma, vitreous cells and haze, vitreous traction bands attaching to the macula or optic nerve, and RD. Leukocoria may be secondary to the granuloma, endophthalmitis, and/or associated RD.
- Children with RDs are often asymptomatic, and thus diagnosis may be delayed until clinical signs become evident. When an RD is central and/or large, leukocoria may be the presenting sign. RDs in children are associated with trauma, ROP, chorioretinal colobomas, high myopia, aphakia, optic nerve pits, Coats’ disease, toxocariasis, and retinal or choroidal tumors.
- Coloboma of the retina and choroid. Colobomas are congenital abnormalities that result from the failure of normal closure of the embryonic fissure. Leukocoria is evident because of white sclera that is visible through the abnormal retina and choroid. Coloboma of the optic nerve, lens, or iris may also be found. RDs may occur due to breaks in the abnormal retinal tissue.
- Myelination of the optic nerve fibers does not normally extend anterior to the lamina cribrosa; however, in approximately 1% of the population, the myelination reaches the retina. This benign condition occurs slightly more often in males and tends to be unilateral. The typical appearance is a feathery white opacity adjacent to the optic nerve and, when dense myelination is present, leukocoria may be evident. Vision is typically normal, although in cases of dense myelination, a corresponding scotoma may be found.
- Retinopathy of prematurity (ROP) is a vasoproliferative retinopathy that occurs in premature and low–birth weight infants. Infants at risk include those born at less than 32 weeks of gestation and/or weighing less than 1,500 to 2,000 g. Diagnosis is typically made at 4 to 6 weeks of age. Leukocoria is because of RD found in the advanced stages of ROP.
The Patient
Clinical Symptoms
Clinical symptoms will vary based on the specific etiology. Parents and/or the patient will complain of a white pupil and possibly reduced vision and/or an eye turn.
Clinical Signs
The clinical signs will vary based on the specific etiology. (See characteristics described earlier.)
Significant History
- Age of onset
- Prematurity and/or low birth weight (LBW)
- Metabolic disorders or other systemic disorders/syndromes
- Maternal infection during pregnancy
- Contact with puppies, eating dirt, or playing in a sandbox
- Positive family history of one of the conditions listed here
Ancillary Tests
A complete ocular examination is necessary and should include measurement of the corneal diameter (microphthalmia), careful examination of the iris for neovascularization (retinoblastoma) and dilated iris vessels (PHPV), examination of the anterior chamber, looking for hyphema or pseudohypopyon (retinoblastoma), evaluation of the lens (cataract, PHPV), and careful anterior vitreous and fundus examination (retinoblastoma, toxocariasis, PHPV, ROP, Coats’ disease, RD, chorio-retinal coloboma, or myelinated nerve fiber).
Additional tests that may be useful in diagnosis include the following:
- B-scan ultrasound (cataract, PHPV, retinoblastoma, RD)
- Serum ELISA test (toxocariasis)
- Anterior chamber paracentesis (toxocariasis)
- Fluorescein angiography (Coats’ disease, retinoblastoma, ROP)
- CT scan and/or MRI of the orbits and brain (Coats’ disease, retinoblastoma)
- Systemic examination (retinoblastoma, congenital cataract)
The Treatment
- Cataracts: Treat any underlying systemic/metabolic conditions. For visually significant cataracts, surgical intervention and aggressive visual rehabilitation are necessary.
- Retinoblastoma: Treatment options include enucleation, external-beam radiation, cryotherapy, laser photocoagulation, brachytherapy, thermotherapy, or chemotherapy.
- Coats’ disease: Laser photocoagulation and/or cryotherapy to the leaking vessels. Surgical intervention is necessary if an RD develops.
- PHPV: Treatment may include cataract extraction and possible fibrovascular vitreal membrane extraction. Aggressive visual rehabilitation should begin after surgery.
- ROP: Treatment options include laser photocoagulation and cryotherapy. Additional surgical intervention is necessary if an RD develops.
- Toxocariasis: Recommended treatment includes oral thiabendazole plus prednisone. Photocoagulation or cryocoagulation may be used to destroy the organism if it is outside the foveal area. Surgery is necessary if tractional RDs occur.
- RD: Laser, cryotherapy, scleral buckling, or vitrectomy is required.
- Coloboma of the retina and choroid: Specific treatment of the coloboma is not necessary; however, if an RD develops, surgical intervention is required.
- Myelinated nerve fiber: No treatment is needed for this benign condition.
Retinopathy of Prematurity
Suzanne M. Wickum
ICD-9: 362.21
 THE DISEASE
THE DISEASE
Retinopathy of prematurity (ROP) is a proliferative vasculopathy affecting premature and primarily low birth weight (LBW) infants. Complications include fibrovascular proliferation and scarring, retinal detachment (RD), vitreous hemorrhage, retinal dragging and macular distortion, leukocoria, myopia, strabismus, and vision impairment/blindness.
- ROP’s prior name was retrolental fibroplasia.
- Annual U.S. incidence of some degree of ROP estimated at 14,000 to 16,000.
- 500 to 600 cases of legal blindness per year.
- Medical advances have enhanced premature infant survival rates but not lowered the incidence of complications.
- The number of severe ROP cases has doubled over the past 5 years.
- Treatment remains suboptimal.
- That ROP can cause devastating vision loss at the very beginning of life underscores the importance of continued research and progress in the implementation of prevention strategies.
Pathophysiology
- ROP development has two phases:
Initial Phase
- Exposure to oxygen after premature birth causes marked constriction of the fragile new retinal vessels. This may represent exaggerated autoregulation, but it compromises retinal perfusion by inhibiting vascular endothelial growth factor (VEGF) and other necessary growth factors.
- VEGF paradox: It is required for normal function but plays an important role in the development of retinovascular disease. Different VEGF isoforms subserve different functions.
- VEGF properties: It stimulates angiogenesis, is a potent inducer of vascular permeability, and is proinflammatory. It is secreted by the retina and retinal pigment epithelium, with increased synthesis induced by ischemia. Conversely, hyperoxia inhibits VEGF synthesis. VEGF has high endothelial cell affinity.
- During the initial phase of ROP, the choroidal circulation plays an important role as, unlike the retinal vessels, the choroidal vessels lack autoregulation and do not constrict in response to elevated oxygen tension. As a result, oxygen moves from the choroidal to the retinal circulation, further aggravating the VEGF inhibition and obliteration of the immature retinal vessels. This may explain why excess oxygen administration exacerbates ROP early on.
- The initial downregulation of VEGF leads to irreversible capillary endothelial cell destruction in affected areas. This results in ischemia of the portion of the retina in the avascular zone and sets the stage for the second phase of ROP development.
Second Phase
- The second phase of ROP development is initiated by the increased metabolic demands of the neural elements of the retina at the same time that marked ischemia has developed in the avascular periphery.
- This results in a marked increase in VEGF production and its effects on angiogenesis and permeability. The severity of this response depends on the extent of avascular retina in any given eye.
- As there has been irreversible destruction of the normal vascular element at the leading edge of the developing retina, the huge upregulation in VEGF stimulates the formation of neovascularization. The growth of these vessels occurs in an uncontrolled fashion.
- This explains why supplemental oxygen may actually be beneficial in this phase, as the oxygen has a downregulating effect on VEGF.
- Selective VEGF blockade will likely play a role in future ROP treatment strategies.
- Cytotoxic effects of oxygen-induced free radicals also play a role in ROP, particularly in very low birth weight (VLBW) infants. Immature retinal vessels lack efficient antioxidative systems.
- If normal retinal vascularization has completed, the vessels are not susceptible to ROP.
Classification of ROP
- The International Classification of Retinopathy of Prematurity (ICROP) was developed in the early 1980s. The scheme is based on the posterior extent of the avascular retina (Zone) and the severity of the retinopathy (Stage). Another important component is the presence or absence of “plus” disease.
Zone
- Zone I: Center is the optic disc. Radius is twice the distance from the disc to the macula.
- Zone II: Larger circle concentric with Zone I. The nasal ora serrata is its nasal border.
- Zone III: Consists of the crescent that Zone II does not encompass temporally (Fig. 21-1).
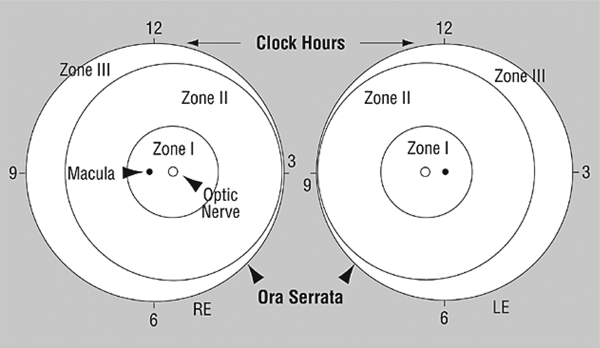
Figure 21-1. Scheme of retina of the right and left eyes showing zone borders, and clock hours used to describe the location and extent of ROP.Data from Committee for the Classification of Retinopathy of Prematurity. Arch Ophthalmol 2005; 123:991–999.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


