1 Pediatric Otolaryngology–Head and Neck Surgery
Overview
1 | What primary germ layers make up the branchial arches, grooves (clefts), and pouches? | Branchial arches and grooves (clefts) are covered externally by ectoderm and composed internally by mesoderm. Each arch has a cartilaginous, muscular, neural, and arterial component. Branchial pouches are composed of endoderm. |
2 | What are the three most common branchial anomalies in order of frequency? | • 70 to 95%: Second branchial arch anomalies dfsdfds sdfssdfsf sfsdfsd sf fsdf dfsfsdf sdfsdfsdfsd • 8 to 10%: First branchial arch anomalies • 3 to 10%: Third and fourth branchial arch anomalies |
3 | What branchial cleft anomaly involves the facial nerve? | First branchial cleft anomaly tracts are close to the parotid gland, particularly the superficial lobe. The tract may pass above, between, or below the branches of the facial nerve. |
4 | What is the second branchial arch structure that normally regresses during development but may be associated with hearing loss and pulsatile tinnitus when present in the adolescent or adult? ( | The stapedial artery |
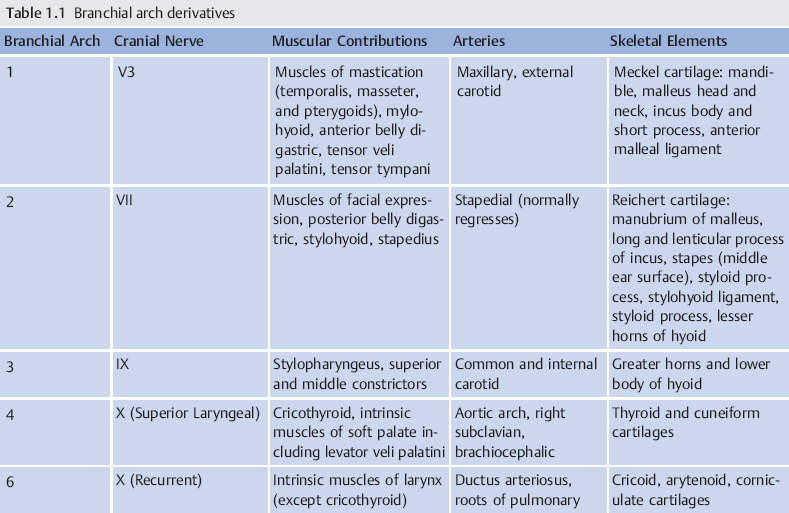
5 | Where is the proximal opening of a second branchial cleft anomaly? | Tonsillar fossa |
6 | What artery can persist in adulthood from the second branchial arch? | The stapedial artery |
What is the course of a persistent stapedial artery? | The stapedial artery rises from the internal carotid artery (ICA), enters the hypotympanum via the Jacobson canal, passes through the crura of the stapes (obturator foramen of the stapes), then passes through the cochleariform process and runs with the tympanic section of the facial nerve before exiting into the extradural intracranial space just before reaching the geniculate ganglion. It replaces the middle meningeal artery, resulting in a hypoplastic or aplastic foramen spinosum. | |
8 | What symptoms are associated with a persistent stapedial artery? | Pulsatile tinnitus, asymptomatic incidental finding, conductive hearing loss (associated stapes ankylosis), sensorineural hearing loss (SNHL), erosion of the otic capsule (rare), and may be associated with additional vascular anomalies (i.e., the ICA) |
9 | Describe the pathway of a third branchial arch anomaly. ( | Piriform sinus of the hypopharynx → through the inferior constrictor muscle medially → greater cornu of the hyoid bone, lateral to the superior laryngeal nerve (nerve of the fourth arch) → over the hypoglossal nerve → inferior to the glossopharyngeal nerve → posterior to the ICA → fistula opens to the skin over the anterior border of the sternocleidomastoid muscle (SCM) |
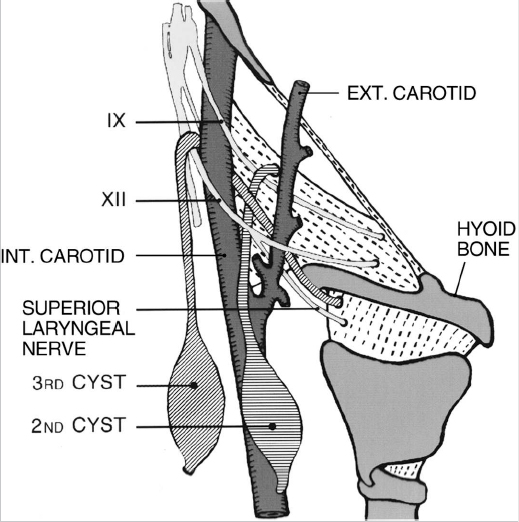
10 | Describe the pathway of a fourth branchial arch anomaly. | Piriform sinus of the hypopharynx → medial to the superior laryngeal nerve (nerve of the fourth arch) → tracheoesophageal groove, parallel to the recurrent laryngeal nerve into the mediastinum → under the aortic arch (left) or subclavian artery (right) (both are fourth arch derivatives) → ascends along posterior surface of the common carotid artery → anterior border of the SCM; it can also follow the common carotid artery to bifurcation → between the ICA and the external carotid artery (ECA) → below the glossopharyngeal → above the hypoglossal → descends inferiorly to exit anterior to the SCM |
11 | What are the clinical presentations for third and fourth branchial cleft anomalies? | Both may be noted as a soft fluctuant mass, abscess, or draining tract located along the anterior border of the SCM. Acute suppurative thyroiditis can be seen. Stridor may be present in newborns with a lateral neck mass. |
12 | What are the typical findings in a patient with branchio-otorenal (BOR) syndrome? | Autosomal dominant syndrome: • Malformed external ears • Preauricular pits • Conductive, sensorineural, or mixed hearing loss • Renal anomalies ranging from mild hypoplasia to complete agenesis |
13 | At which cervical vertebral level is the cricoid cartilage of an infant located? Does this location change as the child grows? | The fourth cervical vertebra The cricoid descends to the level of the seventh cervical vertebra by adulthood. |
14 | Why is the thyroid notch not a palpable landmark for tracheotomy in infants? | Infants have a shortened thyrohyoid membrane, so the hyoid bone is located anterior to the thyroid notch, obscuring the thyroid notch as a landmark for tracheotomy. |
15 | What is the diameter of the subglottis in a full-term infant? | 5 to 7 mm (< 4 mm indicates a subglottic stenosis) |
16 | What are the dimensions of the trachea in a full-term infant? | 4 cm long × 6 mm wide. |
17 | What is the ratio of cartilaginous to membranous trachea? | 4.5:1 |
18 | What additional anomaly should be actively looked for in a patient who has a complete vascular ring? | Vascular sling |
19 | Describe how infants maintain a nasopharyngeal airway while suckling. | The more superior cervical position of the larynx allows overlap of the epiglottis and the soft palate, which allows the flow of milk or formula to be channeled around the dorsum of the tongue and laterally around the epiglottis, thus protecting the airway. |
20 | What is the first paranasal sinus to develop embryologically? | The maxillary sinuses begin developing at 3 weeks of fetal life and are partially pneumatized at birth. They reach full adult size by age 16 years. |
21 | What is the last sinus to undergo pneumatization? | Frontal sinuses Earliest pneumatization occurs at or shortly after age 2 years. |
22 | When do the inner ear structures reach full adult size? | The inner ear structures begin developing at 4 weeks’ gestation and reach adult size by 6 months’ gestation. |
23 | At what age would you expect to see inner ear malformations develop in a fetus? | Between 4 and 13 weeks’ gestation (first trimester) |
24 | When does the auricle achieve the adult form? | ~ 18 weeks gestation. However, it continues to grow in childhood with changes continuing into late adult life. Adult width and length are achieved at different times. Width: age 6 years in females, age 7 in males. Length: 12 in females, 13 in males); 90% of adult size achieved by age 5 |
Orbital size is what percentage of the adult size at birth? | 60 to 65%. This is also the case for the length and width of the cranium. The optic nerve and eye are extensions of the brain and follow brain growth (reaches adult size by 2 or 3 years) rather than growth of the facial skeleton. |
Association Syndromes and Sequences
26 | What anomalies are included in the CHARGE association? | C Coloboma H Heart defect A Atresia, choanal R Retarded growth and development G Genital hypoplasia E Ear anomalies/hearing loss |
27 | What are poor prognostic factors in patients with CHARGE association? | Midline malformations, esophageal atresia, and bilateral choanal atresia |
28 | What head and neck anomalies are related to the CHARGE association? | Choanal atresia, ear abnormalities and hearing loss, facial nerve palsy, pharyngoesophageal dysmotility, laryngomalacia, vocal-cord paralysis or paresis, obstructive sleep apnea, tracheoesophageal fistula, and gastroesophageal reflux. Temporal bone abnormalities, such as hypoplasia of the semicircular canals and Mondini malformation can also occur. |
29 | What gene is involved in the CHARGE association? | CHD7 gene (member of the chromodomain helicase DNA protein family), chromosome 8q12 in 75% of patients with CHARGE association |
30 | What is the incidence of choanal atresia in patients with CHARGE association? | > 65%, > 2/3 bilateral. If unilateral, left > right |
31 | What does VACTERL association stand for? | V Vertebral defects A Anal atresia C Cardiac malformations TE Tracheoesophageal fistula with esophageal atresia R Renal dysplasia L Limb anomalies (most commonly radial anomalies) |
32 | What percentage of patients with VACTERL association have a tracheoesophageal fistula? | 50 to 80% |
33 | What are the major clinical characteristics in patients with velocardiofacial syndrome? | Clefting of the secondary palate, hypernasal speech, pharyngeal hypotonia, structural heart anomalies, dysmorphic facial appearance, slender hands and fingers, and learning disabilities |
34 | What chromosomal anomaly is associated with velocardiofacial syndrome? | About 80 to 100% have a hemizygous deletion of chromosome 22q11. |
35 | What factors lead to velopharyngeal insufficiency in patients with velocardiofacial syndrome? | Cleft palate (occult submucous cleft, overt submucous cleft or soft palate cleft), hypotonia of the pharyngeal muscles, platybasia (an obtuse angulation of the cranial base), and a small adenoid pad |
36 | Why do most patients with velocardiofacial syndrome have chronic otitis media and conductive hearing loss despite having a small adenoid pad? | Abnormal craniofacial anatomy, cleft palate, and associated eustachian tube dysfunction |
37 | What evaluation must be done when performing a pharyngeal flap on a patient with velocardiofacial syndrome? Why? | Nasopharyngoscopy (look for pulsations in the posterior or lateral pharyngeal walls), computed tomography angiography (CTA), or magnetic resonance angiography (MRA); 25 to 30% have medial displacement of their internal carotid arteries. |
38 | What autosomal dominant syndrome is most likely in a child with lower-lip pits, cleft lip, and/or cleft palate? | Van der Woude syndrome |
Describe lower-lip pits in van der Woude syndrome. | Usually bilateral paramedian sinuses in the lower lips placed symmetrically on either side of midline. They can also be median, paramedian, or unilateral (usually left). A single median or paramedian lower lip pit is considered an incomplete expression of the trait. | |
40 | Describe the embryologic formation of lower-lip pits as seen in van der Woude syndrome. | The lower lip of a 32-day embryo consists of four growth centers divided by one median and two lateral grooves. In the 38-day embryo, the lateral grooves disappear unless there is impeded mandibular growth, which results in the formation of a lower lip pit. |
41 | What are the clinical features of Stickler syndrome? | Typical facial characteristics (micrognathia leading to Pierre Robin sequence), hypermobility, and enlargement of joints associated with the onset of arthritis in early adulthood, myopia, retinal detachment, cataracts, and hearing loss |
42 | What is the genetic mutation in Stickler syndrome? | Mutations in the COL2A1, COL9A1, COL11A1, and COL11A2 genes cause Stickler syndrome. These genes are involved in the production of type II, type IX, and type XI collagen, which are components of vitreous, cartilage, and other connective tissues. |
43 | What are the different types of Stickler syndrome, and what are the associated genetic mutations? | • Type 1 (autosomal dominant) are mutations in the COL2A1 gene and are the most common. • Type 2 (autosomal dominant) are mutations in the COL11A1 gene. • Type 3 (autosomal dominant) are mutations in the COL11A2 gene. No ocular abnormalities because COL11A2 is not present in the vitreous. • Type 4 (autosomal recessive) are mutations in COL9A1. |
44 | What are the clinical findings in Pierre Robin sequence? | Micrognathia, glossoptosis, and wide U-shaped cleft palate, leading to upper airway obstruction and feeding difficulties |
45 | Describe the embryology of Pierre Robin sequence. | Arrest in mandibular development at 7 to 11 weeks of gestation (micrognathia) causes the tongue to set abnormally high and posteriorly in the oral cavity (glossoptosis). This prevents fusion of the palatal shelves at week 11 and results in a U-shaped cleft palate. |
46 | What are the most common syndromes associated with Pierre Robin sequence? | • Stickler syndrome (most common) • Treacher Collins syndrome • Velocardiofacial syndrome • Fetal alcohol syndrome • Möbius syndrome • Nager syndrome • Beckwith-Wiedemann syndrome |
47 | How often is Pierre Robin sequence associated with a syndrome? | 80% (20% isolated) |
48 | Discuss the treatment options for upper-airway obstruction in patients with Pierre Robin sequence. | • 70%: Positioning alone (i.e., prone positioning) • 20%: Nasopharyngeal airway, mandibular distraction osteogenesis (gaining popularity), tongue-lip adhesion (being performed less commonly) • 10%: Tracheostomy |
Disorders of the Salivary Gland
49 | Discuss the causes of acute suppurative sialadenitis in premature neonates. | • Reduction in salivary flow • Immunologic immaturity • Presence of bacteria in the oral cavity of neonates • Dehydration • Prolonged orogastric feeding • Congenital anomalies of the floor of the mouth |
What is the treatment for acute suppurative sialadenitis in premature neonates? | Hydration and antimicrobial therapy should lead to a response within 48 to 72 hours. Gland manipulation should be avoided in a preterm child to reduce the risk of systemic septicemia. If no satisfactory improvement is seen, incision and drainage are recommended. | |
51 | What are the causes of pediatric viral sialadenitis? | Epstein-Barr virus, parainfluenza viruses, adenovirus, human herpes virus–6, human immunodeficiency virus (HIV), Coxsackievirus, mumps virus, and influenza virus |
52 | What organ systems are involved in patients with mumps? | Parotid and submandibular glands (diffuse tender enlargement), gonads, pancreas, and meninges |
53 | What is the classic triad of symptoms seen with infectious mononucleosis? | About 80% of patients have the triad of fever, sore throat, and posterior cervical adenopathy that can involve the periparotid or perifacial (submandibular) lymph nodes with subsequent involvement of adjacent glands. |
54 | Discuss the clinical findings of human immunodeficiency virus (HIV)-associated benign lymphoepithelial cysts. | They occur in up to 10% of HIV-positive children, often early in the course of HIV infection with slowly progressive, asymptomatic parotid gland enlargement and often associated with cervical lymphadenopathy. Cysts are usually bilateral (up to 80%), multiple (up to 90%), and involve the superficial lobe. |
55 | What are the most common pathologies causing granulomatous inflammation of the major salivary glands? | Actinomycosis, tuberculosis, atypical mycobacterial infections, and sarcoidosis |
56 | What bacteria are associated with nontuberculous mycobacterial infections of the salivary glands? | Mycobacterium avium-intracellulare (70 to 90% of cases), M. bovis, M. kansasii, and M. scrofulaceum |
57 | What pathology is thought to be a form of sarcoidosis characterized by uveitis, parotid enlargement, and facial paralysis? | Heerfordt syndrome (or uveoparotid fever). Its symptoms include fever, malaise, weakness, nausea, and night sweats. Evaluation includes chest radiography looking for hilar adenopathy and an acetylcholinesterase level. A biopsy from the lip or tail of the parotid may confirm the diagnosis. |
58 | What pathology is characterized by recurrent episodes of nonobstructive, nonsuppurative unilateral (60%) or bilateral (40%) parotid inflammation in a 5-year-old boy? | Juvenile recurrent parotitis (JRP). The peak incidence is between the ages of 3 and 6 years, with predominance in boys. Diagnosis is made on a clinical basis and is confirmed by ultrasonography or sialography, which shows pathognomonic sialectasias (intraductal cystlike dilations). |
59 | What are the medical treatment options for sialorrhea? | • Oral motor training: Exercises are done to encourage swallowing, improve muscle tone and oral sensation, stabilize body and head position, promote jaw stability and lip closure, and decrease tongue thrust. • Behavioral therapy: Verbal and auditory cues can increase the frequency and efficiency of swallowing. • Anticholinergic pharmacotherapy, oral glycopyrrolate, botulinum toxin injections into major salivary glands may decrease the volume of saliva. |
60 | What are the surgical treatment options for sialorrhea? | • Submandibular gland excision* • Submandibular duct rerouting * • Parotid duct rerouting* • Sublingual gland excision • Ligation of parotid ducts • Transtympanic bilateral tympanic plexus neurectomy and bilateral chorda tympani nerve section • Any combination of these procedures *Highest rates of success |
61 | What are the most common benign pediatric tumors of the parotid gland? | Parotid gland hemangiomas constitute 50% of pediatric parotid gland tumors. |
Discuss the development and natural history of pediatric parotid gland hemangiomas. | These hemangiomas may be part of a segmental V3 hemangioma, or they may be isolated focal hemangiomas. They occur at birth or shortly thereafter and act like other hemangiomas, undergoing a rapid proliferative growth phase followed by involution. | |
63 | What is the first-line treatment for parotid hemangiomas? ( | Oral propranolol therapy |
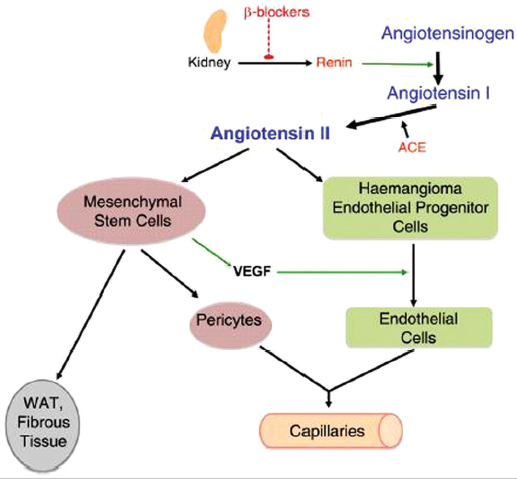
Fig. 1.2 Proposed mechanism of action of propranolol on hemangioma endothelial cell proliferation and differentiation, through blockage of renin production. (Used with permission from Itinteang T, Brasch HD, Tan ST, Day DJ. Journal of Plastic, Reconstructive 64(6):759-765.51.)
Nose and Sinus
64 | What are the two most common maxillofacial fractures in children? | Nasal bone and mandibular fractures |
65 | If a child develops a septal hematoma after sustaining a nasal fracture and subsequently develops a septal abscess, what nasal deformity might the child develop later in life? | Saddle nose deformity, deformed columella or nasal base |
66 | What must be considered in the performance of a complete examination of a child who has sustained a significant nasal injury? | Sedation |
67 | What is the key factor that differentiates the treatment of nasal trauma in pediatric patients compared with adult patients? | Ongoing facial growth |
68 | Why do infants diagnosed with an obstructive septal deviation after nasal trauma require urgent evaluation? | They are obligate nasal breathers. |
69 | When should pediatric nasal fractures be reduced? | Reduction should be done either within 3 to 6 hours of injury, before the onset of swelling, or 3 to 10 days after the injury. If immediate reduction cannot be performed, the fractures should be evaluated 3 to 7 days after the injury, when edema has subsided. |
What is the differential diagnosis for a congenital midline nasal mass? | • Dermoid (most common) • Glioma • Encephalocele • Epidermoid cysts • Hemangiomas • Teratomas • Neurofibromas • Lipomas • Lymphangiomas | |
71 | Name the midline nasal epithelial-lined cyst, sinus, or tract that forms as a result of regression of the embryologic neuroectodermal tract, pulling skin elements into the prenasal space. | Nasal dermoid forms and contains keratin debris, hair follicles, sebaceous glands, and sweat glands. |
72 | What are the clinical findings in a patient with a nasal dermoid cyst? | • It occurs more often in male than in female patients. • Noncompressible mass. • Furstenberg sign is negative (i.e., no enlargement with compression of the jugular veins); it is firm and nontender. • It does not transilluminate. • The cyst is located in the midline, most commonly along the dorsum, resulting in a widened dorsum. • Cyst can be intranasal, extranasal, or intracranial. • It may have a sinus opening with intermittent discharge of sebaceous material. • Hair protrudes through a punctum, is pathognomonic, but not commonly seen. |
73 | How often do nasal dermoids extend intracranially? | In ~ 25% of cases, they most often communicate through the foramen cecum or the cribriform plate to the base of the frontal fossa with extradural adherence to the falx cerebri and are associated with an increased risk of meningitis. |
74 | What radiographic findings suggest a nasal dermoid with intracranial extension? | • Bifid crista galli • Enlarged foramen cecum |
75 | Discuss the treatment of nasal dermoids. | • Treatment involves complete surgical excision. Approaches vary depending on presence of intracranial extension, and recurrence is common. • Extracranial approach: Vertical midline incision, transverse incision, lateral rhinotomy, external rhinoplasty, inverted-U incision, and degloving procedures • Combined approach: Extracranial approach combined with a frontal craniotomy |
76 | What congenital nasal anomaly consists of ectopic glial tissue that lacks a patent cerebrospinal fluid (CSF) communication to the subarachnoid space but in 15 to 20% maintains a fibrous affiliation? | Nasal gliomas |
77 | How do nasal gliomas form? | Theories about formation: • Abnormal closure of fronticulus frontalis, isolating the brain tissue from the intracranial cavity • Nidus of ectopic neuroepithelia • An outgrowth of olfactory tissue through the cribriform plate |
What is the clinical presentation of nasal gliomas? | Extranasal gliomas (60%) are smooth, firm, noncompressible masses that occur most commonly at the glabella, although they may arise along the side of the nose or the nasomaxillary suture line. Intranasal gliomas (30%) are polypoid pale masses that arise from the lateral nasal wall near the middle turbinate and occasionally from the nasal septum and can protrude from the nostril. Combined (10%) Intracranial extension (15%) | |
79 | What congenital nasal anomaly consists of an extracranial herniation of the cranial contents through a defect in the skull? Meninges only? Brain matter and meninges? | Encephalocele, meningocele, and meningoencephalocele, respectively |
80 | How are nasal encephaloceles classified? | Location of the skull base defect: • Sincipital (60%): Arise between the frontal and ethmoid bones at the foramen cecum, immediately anterior to the cribriform plate. • Basal (40%): Arise between the cribriform plate and the superior orbital fissure or posterior clinoid fissure. Note: The most common congenital encephaloceles are occipital (75%). |
81 | What are the subtypes of sincipital encephaloceles? | • Nasofrontal: Defect is located at the glabella between the nasal and frontal bones. • Nasoethmoidal: Sac exits through the foramen cecum, passing under the nasal bones and above the upper lateral cartilages, creating a lateral nasal mass. • Nasoorbital: Sac transverses the foramen cecum before extending into the orbit via a defect in its medial wall. |
82 | What are the subtypes of basal encephaloceles? | • Transethmoidal: Sac extends medial to the superior turbinate via a cribriform plate defect. • Sphenoethmoidal: Sac protrudes into the nasopharynx via a defect between the posterior ethmoid and the anterior sphenoid wall. • Transsphenoidal: Sac also is seen in the nasopharynx, exiting intracranially through an open craniopharyngeal canal. • Sphenoorbital: It protrudes through the superior orbital fissure and out the inferior orbital fissure into the sphenopalatine fossa. |
83 | What are the common clinical findings associated with encephaloceles? | • Bluish/red mass that is soft, compressible • (Positive) Furstenburg test (expands with compression of internal jugular vein) • Pulsatile • Does transilluminate |
84 | What imaging is best for the diagnosis and surgical planning for an encephalocele? | CT and MRI |
85 | Why is surgical repair the treatment of choice for encephaloceles? | To prevent CSF leak, meningitis, and brain herniation |
86 | Describe the four hypotheses that have been offered to explain the development of choanal atresia. | • Buccopharyngeal membrane persistence • Abnormal neural crest cell migration • Bucconasal membrane persistence • Adhesion formation in the nasochoanal region as a result of abnormal mesoderm |
What are the clinical features of choanal atresia? | • 1:5,000 to 8,000 live births • 75% unilateral, right > left • Female-to-male ratio: 2:1 • 50% of patients with unilateral atresia and up to 75% of patients with bilateral atresia have other associated congenital anomalies. • Bony 30%, membranous 70% | |
88 | What are the anatomic features of choanal atresia? | • A narrow nasal cavity • Lateral bony obstruction by the pterygoid plates • Medial obstruction caused by thickening of the vomer • Membranous or bony obstruction |
89 | What syndromes are associated with choanal atresia? | • CHARGE association • FGFR-related craniosynostosis syndromes (e.g., Crouzon syndrome, Pfeiffer syndrome, Apert syndrome, Jackson-Weiss syndrome, Muenke syndrome, Antley-Bixler syndrome) • Down syndrome • Treacher-Collins syndrome • Solitary medianmaxillary central incisor syndrome |
90 | At what age are most infants no longer obligate nasal breathers? | 6 to 9 months |
91 | You are called emergently to evaluate a newborn in respiratory distress. On evaluation, you note cyanotic episodes relieved by crying. Examination is otherwise benign, and the medical history shows no complications. What is the most likely diagnosis? | Bilateral choanal atresia |
92 | How does unilateral choanal atresia commonly manifest? | Typically it is not seen until the patient is aged 5 to 24 months or a young adult as a result of unilateral nasal obstruction or rhinorrhea. |
93 | You suspect possible choanal atresia in a newborn with respiratory distress. What physical examination maneuver can be used to help determine whether choanal atresia is present? | One can attempt to pass a nasogastric tube or small catheter (< 8 French, generally a 5/6 French is used) through the nose. If it does not pass, the presence of choanal atresia is suspected and further workup is required. |
94 | What is the definitive diagnostic test for choanal atresia? | CT scan |
95 | What three surgical approaches can be considered for the treatment of choanal atresia? | • Transpalatal • Transnasal: puncture (Fearon) • Endoscopic endonasal (transnasal, transoral, combined); puncture, drill, dilation |
96 | What symptoms might suggest congenital nasal pyriform aperture stenosis (CNPAS)? | Respiratory distress, poor feeding, failure to thrive, and recurrent cycles of cyanosis and apnea |
97 | What causes CNPAS? | CNPAS occurs secondary to bony overgrowth of the medial nasal process of the maxilla into the nasal aperture resulting in a pyriform aperture smaller than 11 mm. |
98 | What congenital anomalies are associated with CNPAS? | • Holoprosencephaly (HPE): Clinical features include facial dysmorphisms such as ocular hypotelorism, midline cleft lip and/or flat nose, cerebral malformations, learning disabilities, arrhinencephaly, agenesis of the corpus callosum, hypopituitarism, single maxillary central incisor. • Solitary median maxillary central incisor syndrome (SMMCI): Clinical features include severe to mild intellectual disability, congenital heart disease, cleft lip and/or palate and less frequently, microcephaly, hypopituitarism, hypotelorism, convergent strabismus, esophageal and duodenal atresia, cervical hemivertebrae, cervical dermoid, hypothyroidism, scoliosis, absent kidney, micropenis, and ambiguous genitalia. |
What are the treatment options for CNPAS? | • Nonoperative management: Nasal trumpets, topical steroids, and vasoconstrictive drops may be attempted until growth results in increased nasal airway size. • Operative management: A sublabial approach is done to expose the inferior and lateral pyriform aperture. A small diamond burr is then used to widen the bony lateral and inferior margins. | |
100 | What are the features of complete agenesis of the nose (arrhinia)? | • Absence of the external nose, nasal airways, and olfactory apparatus • Hypoplasia of the maxilla • A small high-arched palate • Hypertelorism |
101 | What are the possible causes of congenital anosmia? | • Most commonly autosomal dominant • Defective transportation of odorants to the olfactory neuroepithelium as a result of congenital malformations in the nasal cavity • Disrupted signal transduction or signal propagation • Malformation of regions in the brain essential for olfaction |
102 | What syndromes are associated with congenital anosmia? | • Kallmann syndrome • Congenital insensitivity to pain • Ciliopathies including Bardet-Biedl syndrome and Leber congenital amaurosis |
103 | Where is the most common site of obstruction causing nasolacrimal duct cysts (dacrocystoceles)? | Inferior meatus (membrane of Hasner). Note: Recanalization of the nasolacrimal duct occurs from the lacrimal system inferiorly. |
104 | What symptoms are associated with nasolacrimal duct cysts (dacrocystoceles)? | Epiphora (tearing), nasal obstruction, respiratory distress in neonates (obligate nasal breathers), aspiration, feeding difficulty |
105 | What are common physical examination findings that suggest nasolacrimal duct cyst (dacrocystocele)? | Bluish swelling of the skin overlying the nasolacrimal duct, cyst in the inferior meatus, superior displacement of the medial canthal tendon, and epiphora |
106 | What is the first-line therapy for a nasolacrimal duct cyst? | Massage |
107 | When should you offer surgical intervention for a nasolacrimal duct cyst (dacrocystocele)? | Infant with significant symptoms (i.e., feeding difficulty, infection, or respiratory difficulty) |
108 | What is the surgical treatment of nasolacrimal duct cysts (dacrocystoceles)? | Endoscopic marsupialization (opening the cyst into the inferior meatus). Ophthalmologist may need to probe duct and possibly place stents to ensure patency. |
109 | During week 4 of development, a pouch forms along the dorsal stomodeum. During the week 5, the infundibular stalk and this pouch come into contact and the opening of the pouch is occluded at the buccopharyngeal junction and is separated from the oral cavity by week 6. The pituitary gland then develops from the anterior wall of the pouch (pars distalis) and a small portion of the posterior wall of the pouch (pars intermedia). Normally, the remnant pouch lumen is obliterated; if not, what is this condition called? | Rathke cleft/pouch cyst |
Describe the characteristics of a Rathke pouch cyst. | Non-neoplastic, sellar/suprasellar epithelial lined cyst (sella turcica). Most often they are small and asymptomatic. | |
111 | How do Rathke pouch cysts most commonly manifest? | During the fifth to sixth decade of life, female predominance. They are usually asymptomatic, but large lesions may cause visual disturbance, pituitary dysfunction, and/or headaches. They can be seen on MRI. |
112 | A tumor derived from a Rathke pouch is called what? | Craniopharyngioma |
113 | What benign cyst/bursa can form in the cleavage plane between the nasal cavity and pharynx (Rathke pouch, notochord remnant) as a result of obstruction, inflammation, or infection of the pharyngeal bursa? | Thornwaldt cyst |
114 | What symptoms are most commonly associated with a Thornwaldt cyst? | None. Occasionally patients complain of postnasal drip with intermittent drainage of the cyst or halitosis. If the cyst enlarges or becomes infected, nasal obstruction or eustachian ET dysfunction can result. |
115 | How are Thornwaldt cysts treated? | If the diagnosis is clear, observation and reassurance are recommended. If cysts are dark-colored from hemorrhage or hemosiderin, consider biopsy after obtaining an MRI to rule out intracranial communication. If symptomatic, they can be surgically removed, taking care to remove the entire cysts, which can extend to the prevertebral fascia. |
116 | During a routine examination, you note a smooth, mucus-covered mass within the adenoid pad. Imaging reveals a rhomboid-shaped cyst with no bony or intracranial communication. What is the likely cause? | Intra-adenoidal cyst |
117 | What causes are associated with pediatric sinusitis? | • Immature immune system • Small developing sinuses • Viral upper respiratory infections • Allergy/allergic rhinitis • Immunodeficiency • Gastroesophageal reflux disease • Cystic fibrosis |
118 | What are the diagnostic criteria for pediatric acute bacterial sinusitis? | Clinical diagnosis can be made when a child has an acute upper respiratory infection (URI) and the following (American Academy of Pediatrics [AAP] clinical guidelines, 2013) symptoms: • Persistent illness (i.e., nasal discharge of any quality or daytime cough or both lasting more than 10 days without improvement) or • Worsening course (i.e. new onset of nasal discharge, daytime cough, or fever after initial improvement) or • Severe onset (i.e., concurrent fever with temperature ≥ 39°C or 102.2°F) and purulent nasal discharge for at least 3 consecutive days (Evidence Quality: B, Recommendation) |
119 | What are the predominant pathogens in pediatric acute sinusitis? | Streptococcus pneumoniae (25 to 30%) Haemophilus influenzae (15 to 20%) Moraxella catarrhalis (15 to 20%) |
When should imaging be obtained in the evaluation of child in whom acute bacterial sinusitis is suspected? | Only if there is concern for orbital or intracranial involvement | |
121 | When should a clinician recommend antibiotic therapy instead of supportive care (nasal irrigation, intranasal corticosteroids, topical or oral decongestants, mucolytics, and/or topical or oral antihistamines) and close observation for a child with presumed acute bacterial sinusitis? | AAP clinical practice guidelines, 2013: • Severe onset and worsening course (signs, symptoms, or both) (Quality of Evidence B, Strong Recommendation) or • No improvement after a 3-day course of observation with persistent illness (nasal discharge of any quality or • Cough or both for at least 10 days without improvement) (Quality of Evidence: B, Recommendation) |
122 | What three risk factors are likely to increase the resistance of organisms to amoxicillin in both acute bacterial sinusitis and acute otitis media? | AAP clinical practice guidelines, 2013 • Day care or child care attendance • Antibiotic treatment within the previous 30 days • Age < 2 years |
123 | What antibiotic(s) should be considered for children with acute bacterial sinusitis? | • Amoxicillin ± clavulanate. Duration varies: continue 7 days after resolution of symptoms. • Standard-dose amoxicillin (45 mg/kg daiy divided into two doses): Mild to moderate severity illness in a child who does not attend day care, has not been treated with antibiotics in the previous 30 days, and whose age is > 2 years. • High-dose amoxicillin (80 to 90 mg/kg daily divided in two doses, maximum 2 g/day): In communities with a > 10% incidence of S. pneumoniae resistance • Amoxicillin-clavulanate (amoxicillin 80 to 90 mg/kg daily divided into two doses, maximum 2 g/day): Moderate to severe illness or who attend day care, have received antibiotics within 30 days, or are < 2 years of age. • Ceftriaxone (50 mg/kg dose given intramuscularly (IM) or IV) if unable to tolerate oral administration, followed by an oral antibiotic to complete therapy if improvement is noted within 24 hours. • Clindamycin = cefexime or linezolid and cefexamine or levofloxacin: If the child’s condition worsens after 72 hours on high-dose augmentin • Options to consider if the child is allergic to penicillin: cefdinir, cefuroxime, cefpodoxime, clindamycin and cefexime, linezolid, or a flouroquinolone. • Inpatient IV antibiotics should be considered in pediatric patients with complicated bacterial sinusitis (AAP, 2013). |
124 | Describe the major complications of pediatric sinusitis. | • Orbital/periorbital inflammation from ethmoid sinuses: Preseptal cellulitis, orbital cellulitis, subperiosteal abscess, and orbital abscess • Intracranial spread from the frontal and sphenoid sinuses: Meningitis, epidural abscess, subdural empyema, intra-cerebral abscess, and cavernous or sagittal sinus thrombosis |
125 | How are orbital complications from sinusitis classified? | The Chandler Classification • Group 1: Inflammatory (preseptal) edema of eyelids without tenderness; obstruction of venous drainage; no associated visual loss or limitation of ocular movements • Group 2: Orbital cellulitis with diffuse edema of the adipose tissue in the orbital contents secondary to inflammation and bacterial infections; no abscess formation • Group 3: Subperiosteal abscess, abscess formation between the orbital periosteum and the bony orbital wall. The mass displaces the globe in the opposite direction (usually down and lateral); the proptosis may be severe with decreased ocular mobility and visual acuity. The abscess may rupture into the orbit through the orbital septum. • Group 5: Cavernous sinus thrombosis; progression of the phlebitis into the cavernous sinus and to the opposite side, resulting in bilateral symptoms |
126 | What are the diagnostic criteria for pediatric chronic sinusitis? | American Academy of Otolaryngology (2013) • One or more symptoms of sinusitis > 12 weeks • Six or more episodes of acute sinusitis/year • Acute exacerbations without complete resolution between episodes |
127 | What is the role of intravenous immune serum globulin (IVIG) therapy in pediatric chronic rhinosinusitis? | IVIG may have a role in the treatment of chronic or recurrent acute sinusitis in a select group of patients whose disease is recalcitrant despite maximizing conventional medical therapy. Many believe that IVIG may not be acting as replacement therapy for a humoral immune deficiency but more so as an anti-inflammatory or immune-modulating agent that interrupts the chronic inflammatory process in patients with chronic sinusitis. |
128 | What conventional management options are available for chronic pediatric sinusitis? | Nasal irrigation, intranasal corticosteroids, topical/oral decongestants, mucolytics, and/or topical/oral antihistamines. Patients should also undergo a workup for allergies, reactive airway disease, headache, immuno-deficiency, and cystic fibrosis as indicated. |
129 | Discuss the surgical options for treatment of pediatric chronic sinusitis. | • Adenoidectomy • Maxillary antral lavage • Functional endoscopic sinus surgery: middle meatal antrostomy, anterior or total ethmoidectomy |
130 | What are the indications for sinus surgery for pediatric sinusitis? | Absolute indications: • Complete nasal obstruction in cystic fibrosis attributable to massive polyposis or closure of the nose by medialization of the lateral nasal wall • Antrochoanal polyp • Intracranial complications • Mucoceles and mucopyoceles • Orbital abscess • Traumatic injury in the optic canal (decompression) • Dacryocystorhinitis resulting from sinusitis and resistant to appropriate medical treatment • Fungal sinusitis • Some meningoencephaloceles • Some neoplasms Relative indications: • Chronic rhinosinusitis that persists despite optimal medical management and after exclusion of any systemic disease • Optimal medical management includes 2 to 6 weeks of adequate antibiotics (IV or oral) and treatment of concomitant diseases. |
131 | What primary immunodeficiencies are associated with chronic sinusitis? | • Common variable immunodeficiency • Immunoglobulin (Ig) A deficiency • IgG subclass deficiencies, most commonly IgG3 deficiency (important for defense against Moraxella catarrhalis and Streptococcus pyogenes) |
Discuss the anatomical abnormalities of the sinuses in patients with cystic fibrosis. | The chronic inflammatory disease and decreased ventilation of the sinuses prevent pneumatization, resulting in diminished postnatal growth of the sinus systems already present at birth, the maxillary and ethmoid sinuses. In addition, there is a lack of development, or hypoplasia, of the frontal and sphenoid sinuses. Incidence of nasal polyposis in cystic fibrosis varies from 6 to 48% and does not usually occur before 5 years of age or after age 20. |
Neck Masses and Vascular Anomalies
133 | What is the differential diagnosis for a congenital neck mass? | Lateral neck masses: • Branchial anomaly • Laryngocele • Thymic cyst • Pseudotumor of infancy Midline neck masses: • Thyroglossal duct cyst (most common midline congenital neck mass) • Dermoid cyst • Plunging ranula • Teratoma Entire neck: • Hemangioma • Lymphatic malformation |
134 | What congenital neck mass is most commonly seen in the first 5 years of life as a 1- to 4-cm midline cystic mass that moves cranially with tongue protrusion or swallowing and arises from the foramen cecum? | Thyroglossal duct cyst |
135 | Where in the neck are thyroglossal duct cysts found? | About 60% are located adjacent to the hyoid bone, 24% are between the hyoid bone and base of the tongue, 13% are between the hyoid and pyramidal lobe of the thyroid gland, and the remaining 3% are intralingual. Up to 20% of cysts are slightly off the midline, with a predilection for the left. |
136 | What is the cause of thyroglossal duct cysts? | Thyroglossal duct cysts form as a persistent epithelial tract during the descent of the thyroid from the foramen cecum in the base of the tongue to its final position in the anterior neck. They rarely undergo neoplastic transformation (1%). |
137 | Discuss the evaluation of a child in whom a thyroglossal duct cyst is suspected. | Evaluate for the presence of thyroid tissue in the cyst (45%) and whether there is functional tissue elsewhere: • Ultrasound: Median ectopic thyroid tissue would appear solid on ultrasound. • Thyroid function tests: Patients with median ectopic thyroid tissue are frequently hypothyroid, with elevated thyroid-stimulating hormone (TSH) levels and resultant hypertrophy of the ectopic thyroid tissue. • Thyroid scintiscan: This test is performed if the preceding tests indicate the presence of a median ectopic thyroid to determine whether there is functional thyroid tissue in the cyst and elsewhere. |
138 | What is the procedure for removal of a thyroglossal duct cyst? | Sistrunk procedure: Stresses removal of the central portion of the hyoid bone associated with the thyroglossal duct tract to decrease the risk of recurrence and removal of the tract to the level of the base of tongue. |
What is the clinical presentation of a cervical thymic cyst? | Most occur in the first decade of life as a lateral neck mass, anterior to the SCM, most commonly to the left; 80 to 90% are asymptomatic. However, they may enlarge because of hemorrhage or infection and cause dysphagia, pain, dysphonia, and dyspnea. They are also known to expand with the Valsalva maneuver. | |
140 | What are the possible causes of a cervical thymic cyst? | • Incomplete descent of the thymus into the chest • Sequestration of thymic tissue foci along the descent path into the chest • Failure of the thymopharyngeal duct to involute |
141 | What is the difference between congenital and acquired thymic cysts? | • Congenital : These cysts are usually unilobular and originate from persistent rudiments of the thymopharyngeal duct. They may have epithelium derived from the thyroid and parathyroid glands because of their close association during development. • Acquired : Cysts are multilobular and develop from degenerated Hassall corpuscles (degenerated epithelial cells); they are associated with Sjogren syndrome, apalastic anemia, and acquired immunodeficiency syndrome (AIDS). |
142 | What congenital neck mass is a germ cell tumor made up of ectodermal and mesodermal elements but has no endodermal elements? | A dermoid cyst, which can contain hair follicles, smooth muscle, fibroadipose tissue, and sebaceous glands |
143 | Where do dermoid cysts form? | Along the lines of embryonic fusion |
144 | How are head and neck dermoid cysts categorized? | They are categorized by location: • Periorbital region: Most common in the head and neck; develop along the naso-optic groove between the maxillary and mandibular processes • Nasal dorsum: Develop during ossification of the frontonasal plate • Submentum/floor of mouth: Region of fusion of the first and second branchial arches in the midline, most common location in the neck • Suprasternal, thyroidal, and suboccipital regions |
145 | On clinical examination, how can dermoid cysts be differentiated from thyroglossal duct cysts? | Both most commonly present as painless midline neck masses. Because of their superficial location and lack of mesodermal attachments, dermoid cysts do not move with tongue protrusion or swallowing. Infection of dermoid cysts is also rare because they have no communication with the oropharynx. |
146 | What is the treatment for dermoid cysts? | Surgical excision. If cervical and cannot exclude the presence of a thyroglossal duct cyst, consider a Sistrunk procedure. |
147 | What congenital anomaly arises from embryonic germinal epithelium of all three types: ectoderm, mesoderm, and endoderm? | Teratoma |
148 | What prenatal findings may indicate a cervical teratoma? | Maternal polyhydramnios: Often diagnosed during the prenatal or early neonatal period |
149 | Where do teratomas occur within the head and neck? | Neck (most common), nasopharynx, oropharynx, and oral cavity |
What is the EXIT procedure? | The ex utero intrapartum treatment (EXIT) procedure is a technique used to establish an airway in neonates with airway compression from congenital anomalies diagnosed prenatally. It involves establishing an airway while the fetoplacental circulation is preserved. The fetus is partially delivered via a cesarean section, and the airway is then secured while the fetus remains attached by its umbilical cord to the placenta. | |
151 | What abnormal dilation or herniation of the saccule of the larynx can result in an air- (most often), mucus-, or pus-filled congenital neck mass, respectively? | Laryngocele, laryngomucocele, laryngopyocele, respectively |
152 | What is the difference between an internal and external laryngocele? | • Internal: Dilation lies within the limits of the thyroid cartilage and is seen as cystic swelling of the aryepiglottic fold. • External: Dilation extends beyond the thyroid cartilage in a cephalad direction to protrude through the thyrohyoid membrane. • Combination: These can have internal and external components. |
153 | What benign congenital neck mass presents as a firm, round, nontender mass seen at the junction of the upper and middle third of the SCM that typically presents 2 to 3 weeks after birth? | Pseudotumor of infancy. Also called sternocleidomastoid tumor of infancy, fibromatosis coli, or congenital muscular torticollis. |
154 | What is the natural history of a pseudotumor of infancy? | It is present 2 to 3 weeks after birth, slowly increases in size for 2 to 3 months, and then slowly regresses for 4–8 months; 80 to 100% completely resolve by 12 months. Some benefit is derived from physical therapy. |
155 | What salivary gland is most commonly associated with a plunging ranula? | Sublingual gland |
156 | How do plunging ranulae most often manifest? | Intraoral component appears as a blue dome-shaped lesion in the floor of mouth, usually on either side of the midline. The extraoral portion is seen as a submental mass extending along the inferior border of one side of the mandible. It is not a true cyst. |
157 | What are the treatment options for plunging ranulae? | • Intraoral incision and marsupialization of cyst • Intraoral excision with removal of sublingual gland (preferred) • External cervical approach to identify the cyst, with dissection through the mylohyoid muscle. In combination, excision of the sublingual gland can be performed via an intraoral approach. • OK-432 intralesional sclerotherapy |
158 | How do vascular malformations differ clinically from hemangiomas? | Hemangiomas are typically absent at birth, appear during infancy, undergo rapid growth within the first year of life, and then undergo a variable period of involution. Most vascular malformations are present at birth, demonstrate growth parallel to the child’s development, and do not involute over time. |
How are vascular malformations classified? | Based on the rate of blood flow: • Low-flow lesions: Capillary malformations, venous malformations, lymphatic malformations, and a combined type that has a mixture of either two or three of the low-flow lesions • High-flow lesions: Arterial malformations and arteriovenous malformations | |
160 | What type of malformation is located in the cutaneous superficial vascular plexus, is made up of capillary and postcapillary venules, and grows with the individual, typically becoming darker, nodular, and occasionally leading to hypertrophy of the underlying soft and hard tissues (which can lead to disfigurement)? | Capillary malformation (vascular malformation). Also known as a port-wine stain, it is commonly associated with Sturge-Weber syndrome. |
161 | What syndrome commonly manifests with a triad of facial dermal capillary malformation, ipsilateral central nervous system vascular malformation (leptomeningeal angiomatosis), and vascular malformation of the choroid in the eye associated with glaucoma? | Sturge-Weber syndrome |
162 | How are capillary malformations treated? | The superficial component can be treated with laser photocoagulation with the pulsed-dye laser (585-nm wavelength). Surgery ranging from simple excision of bleeding nodules to wide resection of hypertrophic tissues with soft tissue reconstruction may be used to treat longstanding capillary malformations. |
163 | Describe the manifestation of a capillary malformation. | Also termed port-wine stain, they are the most common vascular malformation, are present at birth, and grow in proportion to body development. They initially appear red and flat and over time may become more purple. They rarely involute. |
164 | What congenital lesion manifests as a bluish discoloration of the skin or mucosa with a soft and compressible deep component, which can swell with Valsalva maneuver or dependent positioning and has no associated thrill or pulse on examination? | Venous malformation |
165 | Where do venous malformations commonly occur in the head and neck? |



