Pediatric Cataract Associated with Ocular and Systemic Anomalies
Rupal H. Trivedi
M. Edward Wilson
As ophthalmic surgeons are well aware, surgery in pediatric eyes can be challenging. Pediatric cataract surgery in eyes with additional ocular anomalies can be even more demanding. In addition, concomitant systemic disease may put a child at higher anesthesia risk (for anesthesia during surgery as well as any future examinations under anesthesia). Visual acuity outcomes may be reduced when the cataract is part of a systemic disease compared with children without a systemic syndrome.1 In this chapter, we discuss pediatric cataracts associated with ocular and systemic anomalies. We do not provide an exhaustive list of anomalies; instead, we review those anomalies where management or outcomes may differ from what would otherwise be expected and if they are not discussed as an individual chapter elsewhere in this book.
ALPORT SYNDROME
Alport syndrome is a hereditary nephritis accompanied by high-tone sensorineural deafness and distinctive ocular signs. It was first reported in the early 1900s (quoted in Ref.2). Guthrie described several cases of familial idiopathic hematuria and suggested maternal genetic transmittance.2 In 1927, Cecil A. Alport described three generations of a family with a combination of progressive hereditary nephritis and deafness. He linked the hematuria with the auditory defects and noted that the severity of the disease corresponded to gender.3 Many families were subsequently described, and the eponym Alport syndrome was coined in 1961. Anomalous basement membranes in the ocular, auditory, and renal systems cause the characteristic triad of abnormalities in patients with Alport syndrome (i.e., ocular signs, sensorineural deafness, and hereditary nephritis).
The ocular signs were initially discussed by Sohar.4 Anterior lenticonus is a distinctive feature, and its presence in any individual is highly suggestive of Alport syndrome. Anterior lenticonus is a rare bilateral condition wherein the anterior surface of the lens protrudes to assume a conical form. Usually, the raised portion consists of clear cortex, while the lens nucleus remains intact and undistorted. Therefore, the deformity is thought to originate in late intrauterine or postnatal life. It is less common than posterior lentiglobus.2 Anterior lenticonus is an important indicator of a poor systemic prognosis because of renal disease in Alport syndrome patients. Anterior lenticonus is more common in male than female patients with Alport syndrome.5 The inheritance is predominantly X linked (85%), although it can be autosomal recessive (10%) or autosomal dominant (5%).6 In addition to Alport syndrome, isolated cases of anterior lenticonus have also been reported, as well as a rare association with Lowe syndrome and Waardenburg syndrome.7
Clinical Features
When using a parallelepiped or optic section during slit-lamp biomicroscopy, the lenticonus is seen as an axial protrusion, often conical, within the pupillary zone of the lens. Minor degrees of lenticonus are difficult to detect but are suggested by a distinctive “oil drop” appearance (effect produced by oil globules in water) of the red reflex on slit-lamp examination. This is due to the fact that none of the rays from the fundus reaches the observer’s eye owing to prismatic reflection in the axial region.1 Examination with a retinoscope can sometimes detect anterior lenticonus even when it is difficult to see with the slit lamp.
Associated Ocular Findings
Refractive Error
Slowly progressing myopia and astigmatism may occur. Literature has reported the use of wavefront sensing to evaluate lenticular irregular astigmatism in eyes with lenticonus.8 The authors noted that irregular astigmatism induced by lenticonus is a relatively symmetrical, spherical-like aberration, in contrast to irregular astigmatism in typical kerato-conus, which is an asymmetrical, comma-like aberration.8
Corneal Abnormalities
Posterior polymorphous dystrophy (PPMD) and arcus juveniles are frequently encountered. Thickening of Descemet layer with later endothelial cell changes can lead to PPMD. It should be noted that certain corneal abnormalities can be observed in renal failure patients regardless of etiology. These include a white limbal girdle of Vogt and band keratopathy.2 Care must be taken to ensure a complete differential diagnosis of the etiology of the patient’s renal disease.
Glaucoma
Iridocorneal adhesions and transparent membranes owing to PPMD result in an increased risk for glaucoma in these eyes.
Cataract
Certain lens opacities are observed in patients with Alport syndrome. First, anterior subcapsular cortical cataracts can occur secondarily to lens capsule rupture of the anterior lenticonus. Spontaneous rupture can lead to a complete white total cataract. On careful inspection, these ruptures are usually preceded by small cracks and splits in the capsule visible at the slit lamp. We now recognize these changes as an impending lens rupture. Second, posterior subcapsular cataracts (PSCs) may appear because of steroid use with post-renal transplant therapy. Third, internal lenticonus may be seen as a posterior lamellar opacity with a posterior projection along the visual axis.5 Combined anterior lenticonus and posterior lentiglobus have been reported.9 Rarely, lens coloboma has been reported associated with Alport syndrome.10
Fundus
Yellow-white to silver flecks within the macular and mid-peripheral regions of the retina can be seen.
Anterior Capsule and Lenticonus
Alport syndrome is caused by a genetic defect within one of the α chains of type IV collagen, a major constituent of basement membranes throughout the body.11 In the eye, it mainly affects the anterior capsule of the lens. Streeten et al.12 identified specific histologic structures that are affected in the crystalline lens capsule. They inferred that the appearance of the lens capsule lesion was similar to the Bowman capsule basement membrane defect in the renal system of Alport syndrome patients. The anterior lens capsule was noted to be one-third the normal thickness centrally and to be more fibrillar than usual, as well as to be associated with large numbers of partial capsular dehiscences containing fibrillar material and vacuoles. The pathologic thinning in eyes with lenticonus as well as the abnormal epithelial cells and fibers may allow bulging of the anterior capsule. Kato et al.11 also noted that the thickness of the anterior lens capsule was decreased, and there were many vertical capsular dehiscences localized in the inner part of the lens capsule. Besides the anatomical problems with the capsule, manipulation of the lens because of accommodation and normal growth causes added stress on the already weakened structure. Abnormal composition of α (IV) chains in the anterior lens capsule of a patient with anterior lenticonus caused by a mutation in the COL4A5 gene has been reported.13
The weakness in the anterior capsule can cause the capsule to rupture, with subsequent formation of an anterior subcapsular cortical cataract or a total white cataract. Traumatic and nontraumatic rupture of the lens capsule has been reported in the literature.14 In contrast, some reports have noted that the anterior capsule is not so fragile (when performing capsulectomy) in these eyes.15,16 These authors theorize that fragility and thinness may present in some patients only in the advanced stages of the disease.15,16
Treatment
Our recommendation for treating an eye with anterior lenticonus is described in Figure 41.1.
Conservative Management
Even if there are no lens opacities, associated refractive errors may affect vision significantly. Glasses and/or contact lenses should be the first line of treatment in such cases. Patients with Alport syndrome should be warned about the possible complication of spontaneous traumatic or nontraumatic rupture of the anterior capsule, leading to total cataract requiring surgery, and informed of the need for prompt evaluation if any sudden change in vision occurs. Careful slit-lamp examinations are needed periodically to look for progression of lenticonus including signs of impending capsule rupture.
Optionally, reduced vision secondary to lenticular changes can be treated with topical mydriatics. Topical phenylephrine can be administered if the patient has axial opacities. If the patient has systemic hypertension, care must be taken when prescribing topical phenylephrine drops, and a diluted concentration can be a viable option.
Surgical Approach
Despite all efforts, conservative management may fail to improve vision satisfactorily. In such cases, clear lens extraction with intraocular lens (IOL) implantation is the reasonable option.15,17 Occasional reports of traumatic and nontraumatic rupture of the anterior capsule have prompted some surgeons to treat this disease more aggressively.9 When signs of impending rupture are present (newly formed cracks and microbreaks in the capsule near the tip of the lenticonus), a clear lens extraction with IOL should be considered. Rupture of the anterior
capsule may require more urgent intervention, and the uncontrolled spontaneously ruptured anterior capsule (often torn from equator to equator) adds intraoperative difficulties.18
capsule may require more urgent intervention, and the uncontrolled spontaneously ruptured anterior capsule (often torn from equator to equator) adds intraoperative difficulties.18
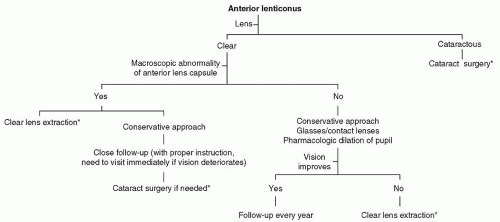 Figure 41.1. Flow diagram showing our current recommendations for treating an eye with lenticonus. *The other eye generally requires surgery within a few weeks. |
Although clear lens extraction may be a viable alternative when treating such eyes, care should be taken to document the corrected distant visual acuity and discuss the options with the patient or parents. As with any elective lens surgery, a rare but severe complication (e.g., endophthalmitis) can occur. Documenting the decisionmaking process is very important in these cases.
Whether operating on a clear lens or a cataractous one, extra care is needed when performing surgery. Herwig et al.19 reported histologic corneal findings in patients with Alport syndrome. Histology revealed marked irregular thickening of the epithelial basement membrane and of Bowman layer and endothelial changes. This suggests that extra care should be taken to protect corneal endothelium during surgery. Extra care is also needed when performing anterior capsulectomy, as the anterior capsule has been noted to be fragile. Some authors, however, have reported that they did not notice any extra difficulty when performing anterior capsulectomy in such cases.20 Probably, anterior capsule fragility is a concern in advanced cases of anterior lenticonus but not when early intervention is attempted. In our experience, the anterior capsulorhexis is not distinguishable from that of other patients of a similar age. Undoubtedly, the center of the capsule is fragile, but if care is taken to control the capsulorhexis peripheral to the fragile center (using ample viscosurgical agent), a strong capsulectomy edge can be created. For the remaining surgical steps, general surgical principles (as outlined elsewhere in this book) should be followed. Once one eye is operated on, the other eye will likely require surgery to achieve better binocular vision and often to prevent the crisis of a ruptured anterior capsule in the fellow eye.
Supportive Treatment
Appropriate genetic counseling is essential for the management of Alport syndrome. Due to the high risk for developmental delay and decreased social integration, management requires a team effort from medical, behavioral, psychosocial, and educational specialists. Patients with Alport syndrome should also consider the use of protective lenses during participation in contact sports (Figs. 41.2, 41.3, 41.4).
Conclusion
To summarize, Alport syndrome offers many challenges to the ophthalmologist. Patients will present with the characteristic triad of hereditary nephritis, hearing loss, and ocular manifestations. A thorough investigation of the hereditary nature of this syndrome within a family is essential. A multidisciplinary approach in the management of these patients, including assistance with developmental and social deficiencies, is necessary to minimize detrimental effects on their quality of life and improve management outcomes. Close follow-up by an ophthalmologist is essential in patients with Alport syndrome. If signs of early or impending anterior lens capsule rupture are observed, clear lens extraction may be considered to avoid an uncontrolled rupture of the anterior lens capsule and subsequent cataract formation necessitating urgent intervention.
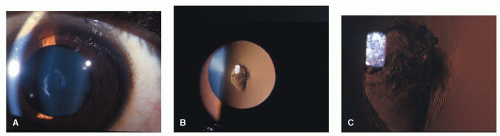 Figure 41.2. Right eye of a 12-year-old African American boy with anterior lenticonus secondary to Alport syndrome. On slit-lamp examination, the paracentral anterior capsule was noted to have findings of early spontaneous rupture. A: Direct split illumination. B: Retroillumination. C: High magnification of spontaneous rupture. |
BONE MARROW TRANSPLANTATION
Cataract is a common late side effect of the radiation and steroid treatments used in bone marrow transplantation (BMT).21 Total dose and duration of corticosteroid therapy are the most important risk factors for development of cataract.22 Total-body irradiation has also been extensively studied as a cataract risk factor.22 Holmstrom et al.23 compared the frequency of cataract development in bone marrow-transplanted children who have been given either total-body irradiation or busulphan as conditioning treatment before BMT. The study confirms total-body irradiation as an etiologic factor for cataract (95%) and suggests that busulphan is also, but less frequently (21%), related to cataract development. The study also shows that cataracts in total-body irradiation-treated children developed earlier after BMT than in busulphan-treated children.23 PSC is the most common type of cataract. PSC typically appears within the first few years after BMT. Frisk et al.24 reported cataract after autologous BMT in children. The authors noted that all children who received total-body irradiation developed PSC (n = 29). Six patients (10 eyes) needed cataract surgery (median time 5 years after BMT, range, 4-9 years). We follow 3 children (6 eyes) in this group. Visual outcome is good in this cohort, as cataract typically develops slowly, bilaterally, and at a relatively older age.
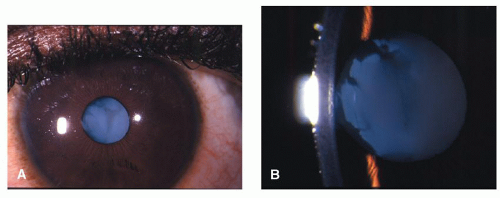 Figure 41.3. The same eye as described in Figure 41.2. Note the total cataract following nontraumatic rupture of the anterior capsule. A: Low magnification. B: Higher magnification. |
CONGENITAL RUBELLA SYNDROME
Congenital rubella syndrome (CRS) remains a prevalent preventable cause of congenital cataract in many parts of the world (Fig. 41.5




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
