3
Pathophysiology of the Blood-Retinal Barrier
Pablo Carnota  Rafael Bueno
Rafael Bueno  Monica Rodriguez-Fontal
Monica Rodriguez-Fontal  John B. Kerrison
John B. Kerrison
INTRODUCTION
In order to maintain the unique microenvironment needed for viability and optimal function of retinal cells, there is a system of dynamic equilibrium adjustments and regulation mechanisms. Akin to the central nervous system, homeostatic regulation is facilitated by a restrictive permeability system known as blood retinal barrier (BRB). Like blood brain barrier (BBB) in brain, BRB restricts the accessibility to the retina of circulating factors such as lipophilic compounds, blood gases, and certain molecules that depend upon active transport.
BRB is known to be disrupted in several pathological situations such as retinal microangiopathies (e.g., diabetic retinopathy), inflammatory-infectious diseases (e.g., Irvine-Gass syndrome) and other exudative conditions (e.g., age-related macular disease (ARMD) or central serous chorioretinopathy (CSC)). This breakdown leads to disturbances of tissue microenvironment, which subsequently accelerates the progression of the pathology (1, 2).
INNER VERSUS OUTER BLOOD RETINAL BARRIER
The BRB is composed of two anatomically and functionally different structures, the inner and the outer BRB. The inner BRB is formed by the endothelial cells of retinal capillaries. The outer BRB is formed by the retinal pigment epithelium (RPE).
Inner Blood Retinal Barrier
Endothelial cells of retinal capillaries display distinctive characteristics in comparison with capillaries in other locations such as lack of fenestrations and presence of apical structures known as tight junctions or zonulae occludentes (Fig. 3-1). These structures not only confer a very restrictive permeability at the intercellular clefts but allow close regulation of transendothelial transport as well as maintenance of endothelial polarity. This phenotypic expression of endothelial cells is dynamic and can be altered, as discussed below, by the local extracellular matrix, soluble growth factors, and heterotypic (other) and homotypic (same) cellular interactions via intercellular junctions. The lumen (5 to 6 μm) of retinal capillaries is created by a single layer of endothelial cells surrounded by intramural pericytes and encompassed by a common basement membrane (3–5). Pericytes have several functions such as regulation of vascular tone, mechanical support of the vessel wall, production of extracellular matrix, and phagocytosis (6, 7). Pericytes exhibit a 1:1 ratio with endothelial cells in normal human retinal capillaries (8), and their pseudopod-like processes encircle the retinal capillary and contain contractile proteins such as smooth muscle type alpha-actin, nonsmooth muscle actin and smooth muscle myosin (6, 9) (Fig. 3-2). Additionally, pericytes release extracellular components, such as fibronectin, which provide anchorage points for mechanical force transfer during capillary lumen constriction (6).
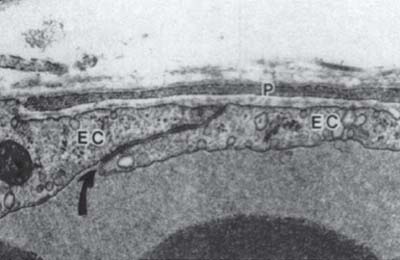
Figure 3-1. Electron micrograph of a control venule showing normal ultrastructural appearance of a pericyte process (P), endothelial cells (EC) and an endothelial cell junction (curved arrow). The space between the pericyte and endothelial cells is occupied by basal lamina. x31,000. (With permission from Shepro D, Morel N. Pericyte physiology. FASEB J 1993;7: 1031–1038).
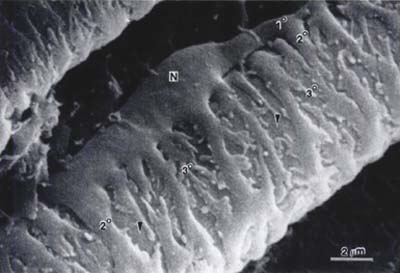
Figure 3-2. Scanning electron micrograph of pericytes covering the surface of an arterial capillary of the rat mirabile. Note the plump nuclear area (N), a primary process (1∘) of the pericyte paralleling the long axis of the capillary, secondary processes (2∘) encircling the capillary at right angles to the primary process, and branching, tertiary processes extending from the secondary process. The surface of an underlying endothelial cell can be seen at the arrowheads. x8804. (From Shepro D, Morel N. Pericyte physiology. FASEB J 1993;7:1031–1038.)
Tight junctions are formed by a complex network of proteins that importantly can be regulated and dynamically altered by different conditions and pathologies. There are three types of transmembrane proteins within tight junctions: occludins, claudins, and junctional adhesion molecules (JAM). The transmembrane proteins bind junctional proteins in adjacent cells and create the actual barrier. Other proteins have been identified such as intracellular protein ZO-1 and cadherin, the main component of another type of junction between cells known as adherens junction.
Occludin was the first identified membrane component of tight junctions (10) (Fig. 3-3). However, subsequent studies demonstrated that occludin was not essential for the tight junction assembly and rather was involved in signal transduction of endothelial cells (11, 12). Actually, other studies suggested that the main role of occludin could be the control of paracellular flux, mainly of ions and small organic cations (13–15). Thus, occludin contributes both structurally and as signal transducers and regulators of barrier properties. Two studies on diabetic retinas and on bovine retinal endothelial cells treated with vascular endothelial growth factor (VEGF) have revealed decreased occludin content concomitant with increased BRB permeability (16, 17). Another protein, tricellulin, has recently been identified as a protein localized specifically at regions where three cells make contact (18). Together, these proteins create the final subset of known transmembrane tight junction proteins.
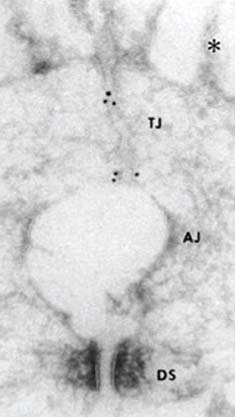
Figure 3-3. Ultrastructural localization of occludin. Ultrathin cryosection of formalin-fixed intestinal epithelial cells were labelled with monoclonal antibodies anti-occludin-2. Gold particles accumulated at the tight junction region (TJ), and are hardly detected in the adherens junction (AJ) and desmosome (DS) regions. AJ is artifactually opened, probably due to the formaldehyde fixation. (From Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 1993;123:1777–1788.)
Later, claudins were found to be other components of tight junctions (19) (Fig. 3-4). Accumulative evidence has revealed that they are key molecules in the tight junction assembly by playing a very important role in the control of ion flux (20–25). However, the complexity of the system is only beginning to be understood. Claudins are a multigene family of more than 20 members (26–28), and the pattern of expression of different claudin family members varies among tissue types, which confers tissue-specific properties to tight junctions (20). Claudins expressed in endothelial cells of neural (and retinal) tissue are claudin-1, claudin-3, claudin-5, and claudin-12 (28–30). They are suggested to be candidate molecules responsible for structural endothelial barrier function as shown by a study of claudin-5-deficient mice that disclosed that claudin-5 is indispensable for the barrier function of neural blood vessels for small molecules (29). Alterations in claudins have been associated with a variety of disease states (31–32). Mutations in claudin 16 exemplify the role of claudins in barrier selectivity since it has been demonstrated as the cause of familial hypomagnesemia with hypocalcemia and nephrocalcinosis (33).
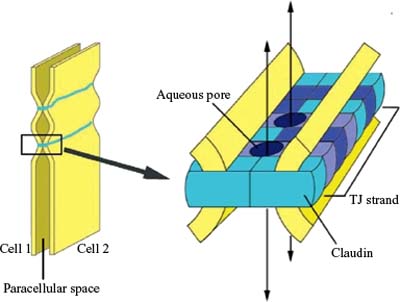
Figure 3-4. Model for the assembly of claudins into tight junctions (TJ) strands. A TJ strand of cell 1 associates laterally with a TJ strand in the opposing membrane of the adjacent cell 2 to constitute a paired TJ strand. Different claudin species, which are indicated in different colors, copolymerize in individual TJ strands, and associate between adjacent cells in both a heterotypic and homotypic manner. Aqueous pores are postulated to occur within paired TJ strands. (From Furuse M, Tsukita S. Claudins in occluding junctions of humans and flies. Trends Cell Biol 2006;16: 181–188.)
Another integral membrane protein of tight junctions is JAM found at the tight junctions of epithelial and endothelial cells. The exact function of JAM is still unclear. It has been demonstrated that transfection of JAM into epithelial cells does not reconstitute tight junctions strands (34). However, it contributes to tight junctions assembly and cell-to-cell adhesion in epithelial and endothelial cells (35–38) and contributes to extravasation of monocytes through endothelial monolayers (38).
Zonula occludens or ZO proteins are intracellular proteins associated with the cytoplasmic surface of tight junctions. The first ZO protein recognized, ZO-1, is a transmembrane protein that acts as a scaffold due to its multiple binding domains; its several domains bind to transmembrane tight junction proteins occludin, claudin, and ZO-2 and to adherens junction proteins afadin and cadherin (39–46) (Fig. 3-5). ZO-1 presence can readily be observed in both retinal vascular endothelial cells and RPE. In both cell types, agents that induce permeability such as VEGF or hepatocyte growth factor induce redistribution of ZO-1 from the cell border to the cell interior (47, 48). In contrast, increasing barrier properties increases ZO-1 border staining. Elevated hydrostatic pressure across bovine aortic endothelial cells first increases water and solute flux, which then decreases over the next 30 to 60 minutes, reaching a new equilibrium; ZO-1 can readily be seen to increase at plasma membrane during this adaptive response (49, 50). Therefore ZO-1 acts as a central organizer of the tight junction complex.
There is another type of junction between endothelial cells called adherens junction. Adherens junctions are characterized by the expression of cadherins (Ca++-dependent transmembrane adhesion proteins) whose function consists of maintaining a space between cell membranes, while allowing homotypic cell contact (51).
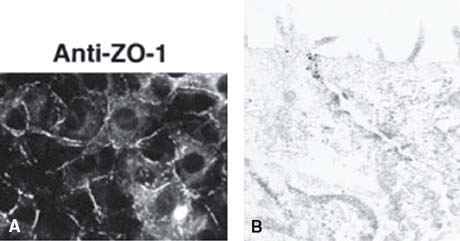
Figure 3-5. Localization of ZO-1. A: Cells stained with a monoclonal antibody against ZO-1 showing a subcellular localization of this protein. (From Imamura Y, Itoh M, Maeno Y, et al. Functional domains of alpha-catenin required for the strong state of cadherin-based cell adhesion. J Cell Biol 1999;144:1311–1322.) B: Immunoelectron micrograph of the junctional complex region in cells stained with a monoclonal antibody against ZO-1; ZO-1 labelling is exclusively concentrated in the junctional complex region. (From Yamamoto T, Harada N, Kano K, Taya et al. The Ras target AF-6 interacts with ZO-1 and serves as a peripheral component of tight junctions in epithelial cells. J Cell Biol 1997;139:785–795.)
Within this microenvironment several soluble vasoactive molecules have been identified as playing a more or less important role in the regulation of permeability of the inner BRB. Molecules such as nitric oxide, adenosine or CO2 or hypoxia cause retinal pericytes relaxation whereas other molecules such endothelin-1, angiotensin II, ATP or hyperoxia lead to pericyte contraction.
Tissue oxygen concentration is known to influence the expression of various molecules including growth factors, cytokines, and enzymes (52). Hypoxia stimulates cellular production of erythropoietin (53), VEGF (54), tyrosine hydroxylase (55), phosphoglycerate kinase 1, and lactate dehydrogenase A (56). Many of these hypoxia-sensitive molecules are implicated in adaptive processes of organisms to hypoxic circumstances such as erythropoiesis, angiogenesis, increase in respiratory volume and conversion of the metabolism to an anaerobic state. In addition, changes in expression of certain oxygen-sensitive molecules can also trigger the progression of disease processes in patients with cerebral ischemic diseases, diabetic retinopathy and other diseases (57). Among components of tight junctions, occludin, claudin-1, claudin-3 and claudin-5 are reported to be hypoxia-sensitive molecules and promote breakdown of the neural endothelial barrier (52, 58, 59).
Endothelial cells synthesize nitric oxide in response to several stimuli like increased local shear force, bradykinins, insulin-like growth factor 1, acetylcholine, thrombin and various platelet products (60–62). Nitric oxide diffuses to the pericyte and smooth muscle cell surface, where it binds to guanylate cyclase, causing intracellular cGMP accumulation and vasodilation (63, 64). In addition to its vasodilatory actions, nitric oxide protects vessels by inhibiting platelet aggregation, platelet granule secretion, leukocyte adhesion, and possibly smooth muscle cell proliferation (65). However, high levels of nitric oxide can induce retinal toxicity through neuronal nitric oxide synthase, which may be important during retinal ischemia, degenerative retinal disease, and diabetic vascular dysfunction (66–71).
Endothelins are released by endothelial cells on the abluminal side of capillary and bind to receptors on adjacent pericytes and smooth muscle cells (72). Endothelins (ET1, ET2, and ET3) are the most potent vasoconstrictor agents currently known (73). ET-dependent contraction is mediated by three receptor subtypes (ETR A, B, and C) (74). Human studies have shown that vasoconstriction following intravenous injection of ET1 is mediated through ETR-A (75). In general, ETR-A mRNA is localized to pericytes and smooth muscle cells, whereas ETR-B mRNA can be found also in endothelial cells. Animal studies have shown that stimulation with ET1 on pericytes causes contraction, aggregation of actin fibers, and proliferation (76–78).
Vascular endothelium generates basal levels of superoxide anions but can be stimulated to produce deleterious amounts of these molecules during many pathologic conditions (79). Superoxide anions inactivate nitric oxide, thereby inhibiting vasodilatory capacity. This pathologic inhibition may mediate vascular changes found in hypertension, hypercholesterolemia, retinopathy of prematurity, diabetes, and ischemia/reperfusion injury (79, 80). Superoxide anions may induce endothelial damage resulting in a prothrombotic state; this pathologic mechanism may contribute to diabetic retinopathy (81).
VEGF stimulates occludin phosphorylation through activation of the protein kinase C (PKC) signaling pathway. Binding of VEGF to its tyrosine kinase receptor activates PKC signal transduction pathway and this activation has been correlated to VEGF’s mitogenic effects in endothelial cells (82). In a recent study on bovine retinal epithelium cells, Harhaj et al. showed that VEGF stimulates occludin phosphorylation increasing the endothelial permeability, an effect that can be inhibited using PKC inhibitors or by expressing a dominant negative PKC-bII mutant (83). However, only part of VEGF-induced permeability was prevented by classical PKC inhibitors indicating additional pathways are likely involved in the regulation of permeability.
Another regulator of vascular permeability is the renin-angiotensin system. Human and animal ocular tissues locally express the necessary components (renin, angiotensinogen and angiotensin-converting enzyme, ACE) for an intrinsic renin-angiotensin system (84–88). ACE bound to endothelial cell luminal surface rapidly converts angiotensin I to angiotensin II; ACE itself inactivates bradykinin and angiotensin II stimulates retinal vasoconstriction via smooth muscle cells and pericytes (89). Isolated porcine ophthalmic arteries vasoconstrict in response to angiotensin II and relax in response to bradykinin via a nitric oxide-dependent mechanism. ACE-inhibitors used in this model prevented angiotensin-mediated constriction and potentiated bradykinin relaxation (90).
Vascular permeability often accompanies angiogenesis, particularly in pathologies of the BBB and BRB. New evidence suggests that tight junction proteins may contribute to growth control through either activation or sequestration of signaling components, potentially linking changes in permeability with angiogenesis. However, most studies have focused on epithelial to mesenchymal transition and not on vascular endothelial cells.
Outer Blood Retinal Barrier
The main component of outer BRB is the RPE. The RPE prevents blood components from passing freely to the subretinal space from fenestrated blood vessel of the choriocapillaris. Tight junctions between RPE cells (similar to endothelial cells in the inner barrier) are the principal mechanism involved in the barrier. However, tight junctions of RPE are more permeable than inner BRB, emphasizing the differential physiological role of the outer and inner BRB. Apical and basal RPE membranes contain a variety of selective ion channels and a variety of active and facilitative transport systems that control the movement of ions and water and the transport of metabolites, such as glucose and amino acids, from choriocapillaris to neural retina and vice versa (91). There are different channels and transporters within apical and basal surfaces, such as an electrogenic sodium-potassium pump on apical membrane and a chloride-bicarbonate exchange transporter on basal membrane. This asymmetry between apical and basal membranes is allowed by tight junctions and accounts for the development of a voltage (called the standing potential) across the RPE that results in a net movement of ions away from the subretinal space that drives the transport of water towards the choroid. Passive transport mechanisms, such as intraocular pressure against the retina and osmotic pressure from the choroid, also tend to drive fluid out of the subretinal space, but passive water movement is normally restricted by the BRB. When the RPE barrier is damaged, fluid actually leaves the subretinal space faster than under normal conditions. In other words, a normal eye will not accumulate fluid in the subretinal space just because the RPE barrier is disrupted (92). These observations have relevance for clinical disorders such as serous detachments. They show that the presence of a defect in the RPE is not by itself sufficient to cause serous detachment, even though it may be necessary (93) (see further discussion below in pathophysiology of CSC).
IMMUNE PRIVILEGE
The eye is an immune privileged site, protected from the immune system by different mechanisms (94). At the level of posterior segment of the eye, RPE cells have a crucial role in induction and maintenance of this immune privilege (95). Failure in outer BRB leads to a breakdown of the immune privilege with subsequent development of intraocular inflammation, as happens in posterior uveitis.
Much work has been done to better understand mechanisms underlying RPE protection role, especially how these cells interact with lymphocytes. It is now evident that active processes are likely to be involved. Liverdsidge et al. first showed that RPE cells could inhibit lymphocyte proliferation (96). From then on, three main pathways have been proposed to explain this RPE immunosuppression. First, RPE cells might inhibit T-cell proliferation through secretion of inhibitory factors like prostaglandins and immunosuppressive cytokines. Among the latter, transforming growth factor b or IL-1 receptor antagonist have been proposed (95–96). Second, the apoptosis phenomenon might also contribute to RPE induced immunosuppression. Apoptosis is a programmed cell death with no inflammatory reaction commonly used by the immune system to eliminate cells (opposite to processes such as necrosis). Moreover, apoptotic body phagocytosis has been shown to downregulate proinflammatory cytokine secretion in macrophages (97). Interestingly, several studies have demonstrated that apoptosis occurs within T-cells following coincubation with RPE cells, suggesting that this could be an important mechanism in RPE immunosuppression (98, 99). Finally, other studies have shown that, by lacking expression of a complete set of costimulatory molecules, RPE cells only partially activate lymphocytes and act as deviant antigen presenting cells, driving the lymphocyte to a state of hyporesponsiveness rather than activation (100–102).
Considering that human RPE cells are known as phagocytic cells, since they largely phagocytose photoreceptor outer segments, phagocytosis may also contribute to RPE immunosuppression directly by decreasing T-cell number and clearing apoptotic lymphocytes and indirectly by downregulating pro-inflammatory cytokine secretion such IL-1b. Therefore, they participate in vivo in induction and maintenance of the immune privilege of the eye, preventing the development of intraocular inflammation (100).
PATHOPHYSIOLOGIC MECHANISMS OF BREAKDOWN OF THE BLOOD RETINAL BARRIER
Diabetic Retinopathy
It is known that fundus changes observed in nonproliferative diabetic retinopathy are the result of pathologic changes at the level of the retinal blood vessels, but the exact mechanism that leads to the breakdown of inner BRB remains poorly understood. The choroidal vasculature is also altered in diabetic patients, but not as significantly as retinal vasculature. Disruption of inner BRB is an early phenomenon in preclinical diabetic retinopathy (PCDR). Two vascular permeability pathways may be affected: the paracellular pathway involving endothelial cell tight junctions and the endothelial transcellular pathway mediated by endocytotic vesicles (103, 104). The relative contribution of both pathways to vascular permeability in PCDR is still unknown (105).
The breakdown of inner BRB is a consequence of retinal ischemia in diabetic retinopathy, and there is a hypothesis about how ischemia develops in this disease. Several histopathological changes are known to occur in diabetic retinopathy in humans and in animal models: pericytes and endothelial cell loss, capillary dropout, and basement membrane thickening. As commented above, in normal retinal vasculature, endothelial cells and intramural pericytes are distributed in a ratio of approximately 1:1. It has been shown that eyes with diabetic retinopathy demonstrate a preferential loss of microvascular pericytes (106) (Fig. 3-6). This happens even before retinopathy becomes clinically apparent. Lack of pericytes in retinal capillaries leads to loss of retinal blood flow autoregulation in response to changes in partial pressure of oxygen and to a functional alteration in endothelial cells. As retinopathy progresses, and even later than appearance of microaneurysms, diabetic retinal capillaries can become acellular (with no endothelial cells). A study by de Venecia et al. provided direct evidence that these acellular ghost capillaries are not perfused in life (107). Perhaps as a response to the retinal hypoxia caused by this capillary dropout, the adjacent capillary bed becomes dilated and hypercellular. Subsequently the lumen of these capillaries narrows and leads, along with other contributing factors such as thickening of endothelial basement membranes and blood viscosity abnormalities, to more retinal capillary closure.
The sorbitol pathway could be an explanation for the pericyte and endothelial dropout and the vascular basement membrane thickening in these patients. Under hyperglycemic conditions, aldose reductase reduces glucose to its sugar alcohol, the sorbitol, which can subsequently reach high levels in cells, and is toxic to them. Besides this, hyperglycemic retinal pericytes display blunted ability to respond to ET1, via PKC-induced downregulation of ET receptors (108–110), which leads to alteration in the retinal blood flow autoregulation. Osmotic swelling of the endothelium secondary to intracellular sorbitol accumulation could also impair blood flow through small vessels by narrowing their lumens. However, Akagi et al. found that aldose reductase is present only in pericytes but not in endothelial cells, which explains the specific loss of pericytes in diabetic retinopathy; the loss of endothelial cells, therefore, could be secondary to loss of pericytes (111). Several studies postulate that the sorbitol pathway is also the mechanism for vascular basement membrane thickening although its mechanism remains unclear (112–116). However, the role of aldose reductase inhibitors (sorbinil) in preventing these microvascular changes in animal models has been controversial and their efficacy in retarding the progression of diabetic retinopathy in humans has not yet been demonstrated (113–117).
Defects in normal tight junctions between endothelial cells may arise secondary to obstruction of adjacent vessels or from the dropout of capillary pericytes. Even before any retinopathy is demonstrable clinically, evidence of abnormal vascular permeability may be seen on fluorescein angiography as focal staining of arteriole wall. As retinopathy develops and progresses, accumulation of fluid and plasma constituents in the surrounding retina can lead to marked retinal thickening, cystoids changes in the fovea and formation of hard exudates (118). Moreover, this process could explain the link between abnormal retinal hemodynamics (such as arterial hypertension) and the prevalence of macular edema (119, 120).
Besides the paracellular pathway there is a transcellular pathway that contributes to breakdown of the inner BRB, and it has two possible anatomic explanations. One, retinal endothelial cells, normally unfenestrated, develop fenestrae similar to those in choriocapillaris, allowing fluid and plasma components to go out of capillaries (121). The other potential explanation is an increase in transport by endocytic vesicles (122), but this mechanism has not yet been reported from human or experimental animal subjects with diabetes.
There are other pathogenic mechanisms by which capillary nonperfusion occurs and that contribute to inner BRB breakdown.
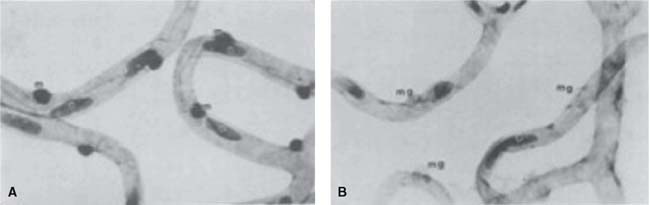
Figure 3-6. Human trypsin-digested retinal capillaries stained with PAS and hematoxylin. The normal distribution of endothelial cells (e) and mural cells (m) are illustrated in (A), while in (B), the selective loss of these mural cells (mural cell ghosts, mg) in the diabetic retina can be observed. (From Akagi Y, Kador PF, Kuwabara T et al. Aldose reductase localization in human retinal mural cells. Invest Ophthalmol Vis Sci 1983;24:1516–1519.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


