Pathology of the Cornea-Sclera
Henry D. Perry
J. Douglas Cameron
CONGENITAL ABNORMALITIES OF THE CORNEA
Developmental abnormalities of the cornea are usually associated with other ocular and nonocular abnormalities because the cornea develops in conjunction with several germinal layers. Surface ectoderm develops into corneal epithelium, Bowman’s layer, and crystalline lens. Anomalies arising from defects of surface ectoderm range from cryptophthalmos to cataracts.1 Neural crest mesenchyme develops into the corneal stroma, the anterior chamber angle filtering apparatus, and the iris stroma. Associated anomalies of neural crest mesenchyme range from a localized faint corneal stromal opacity to major alterations of the anterior chamber filtering mechanism (Axenfeld’s anomaly) and congenital glaucoma. Neuroectoderm develops into the iris pigment epithelium. Abnormalities of the optic cup may lead to such anomalies as microcornea and megalocornea and possibly macular hypoplasia.2,3 Because the cornea develops at an early embryonic stage, coexisting abnormalities in other organ systems are to be expected. Facial, dental, genitourinary, cardiac, and skeletal abnormalities commonly are associated with corneal defects.4,5,6,7 Many types of pathologic processes may lead to developmental abnormalities of the cornea that are indistinguishable clinically from one another. Congenital corneal opacities have been associated with the connective tissue disorder Ehlers-Danlos syndrome,8 with congenital infection,9 or as a manifestation of a recognized teratology.10,11 Some types of processes, such as corneal keloid, may be expressed long after birth.12
No all-encompassing system of classification exists for developmental abnormalities of the cornea. Classifications of appearance and shape are not mutually exclusive (e.g., sclerocornea often is associated with cornea plana and vice-versa). Classification by neural crest origin is appropriate for some elements of the cornea (e.g., the endothelium) but not for all components of the cornea.13 Classification by direct cause on a biochemical level is becoming known for some of the genetically determined corneal diseases (formerly known as corneal dystrophies). The ultimate classification will be based on gene locus abnormalities responsible either for molecules of abnormal tissue induction or for abnormal tissue function.
AGENESIS OF THE CORNEA
Total absence of the cornea as an isolated finding without major abnormalities of the surrounding structures has not been reported.
Cryptophthalmos is a rare condition in which the eyelids fail to form. The surface ectoderm component of the cornea is absent in cryptophthalmos; however, the neural crest components are present and make up the external surface of the globe and the anterior segment, even though these tissues may not be normal.14
Cryptophthalmos may present as an isolated abnormality or may be one of a spectrum of congenital abnormalities. Fraser syndrome is an autosomal recessive multiple malformation syndrome whose major manifestations are cryptophthalmos, syndactyly, laryngeal atresia, and urogenital defects.15,16,17 Cryptophthalmos is found in 93% of cases. In Fraser syndrome there is a stillborn rate of 26% and a first year mortality of 19%.18
ABNORMALITIES OF SIZE
Microcornea
Microcornea exists when the largest corneal diameter is less than 10 mm.19 The globe in microcornea is normal in size.5 This is in contrast to microphthalmia, in which the cornea and the globe are both small. Microcornea may be entirely normal in clinical appearance or may be associated with sclerocornea.20 Associated ocular findings include cataract, corectopia, high myopia, macular hypoplasia,3 and nystagmus. Although microcornea rarely is associated with congenital glaucoma, approximately 20% of individuals who have microcornea will develop complex mechanism glaucoma later in life.21 Associated systemic conditions include skeletal abnormalities,22 Weill-Marchesani syndrome, Ehrlos-Danlos syndrome,23 and Norrie’s disease. The inheritance pattern of microcornea is autosomal dominant or X-linked.1,20,24 Histologically, the cornea is normal.
Megalocornea
Megalocornea exists when the largest corneal diameter is greater than 13 mm (Fig. 1).19,25 Megalocornea is a primary overgrowth rather than a secondary distention of the cornea.
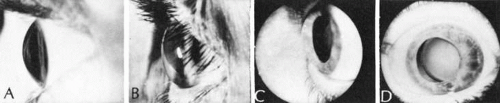 Fig. 1. Megalocornea. A. The corneal diameter measures 15 mm. The remainder of the eye is normal. B. Megaloglobus consists of an enlarged cornea and globe. C. Megalocornea in the brother of the patient shown in A (a third brother also had megalocornea). The corneal diameter measures 16 mm. D. The crystalline lens is dislocated, and chronic open angle glaucoma is present. (Courtesy of SEI Photoarchives.) |
The condition is not progressive. The cornea clinically is normal, although associated prominent iris processes and a heavily pigmented trabecular meshwork may occur. Myopia, high astigmatism, anterior embryotoxon, Krukenberg’s spindles, a pigmentary-like glaucoma, and cataract also have been found in association with megalocornea. The lens may dislocate later in life and cause secondary glaucoma. Associated systemic conditions include osteogenesis imperfecta, Marfan syndrome, and Alport’s syndrome. The inheritance pattern most commonly found is sex-linked recessive (Xq21.3-q22 region)26 although autosomal dominant and autosomal recessive cases have been identified.
Histologically, megalocornea shows a normal-sized cornea but an increased length and thickness from the end of Bowman’s membrane to sclera (limbal region). Endothelial cell density by specular microscopy is normal. An increased endothelial cell population suggests a process of total corneal hyperplasia.27
ABNORMALITIES OF CURVATURE
Cornea Plana
Cornea plana is an abnormal flattening of the curvature of the cornea that decreases the refractive power or the cornea. This is a rare condition occurring worldwide with a high prevalence in Finland.28,29 The genetic defect has been mapped to the long arm of chromosome 12 for both the mild dominantly inherited form and the more severe autosomal recessive form.30,31 Clinical findings among those more severely affected include a greatly reduced corneal refraction (25–35 D) causing high hyperopia, slight microcornea, an extended limbus zone, a central deep corneal opacity, and marked arcus senilis, even before the age of 20 years.29 Mutations in keratocan (KERA), a small leucine-rich proteoglycan, have been shown to be responsible for cases of autosomal recessive cornea plana.32,33,34
The cornea generally is normal in diameter. Cornea plana often is associated with sclerocornea and may be associated with microcornea, posterior embryotoxon, congenital cataract, iris and ciliary body coloboma, and macular aplasia. Diffuse, deep stromal opacities may be present. The anterior chamber may be shallow, and the upper lid may appear ptotic. Astigmatism usually is present, although the eyes may be either myopic or hyperopic. The corneal tissue histologically is normal. The axial length of eyes that contain cornea plana is normal.35 By in vivo confocal microscopy the thickness of the epithelium is observed to be reduced and Bowman’s membrane is absent. The overall corneal thickness was normal or slightly reduced; however, the cytology of the keratocytes is abnormal, as is the appearance of the subbasal nerve plexus. Backscattering of light is noted. The epithelial and endothelial cell morphology is normal.36
Keratoglobus
Keratoglobus is a bilateral abnormal steepening of a uniformly thin cornea that is generally normal in diameter. Clinically the cornea has a protruding, globular appearance. The condition usually is stable and asymptomatic except for isolated instances of spontaneous ruptures of Descemet’s membrane.37 Glaucoma, cataracts, and lens dislocations are not associated with keratoglobus as they are with megalocornea. No definite inheritance pattern has been noted for keratoglobus.
The corneal stroma is approximately one-third normal thickness except at the periphery, where it approaches normal thickness. The corneal epithelium is diffusely thin. Defects may occur in Bowman’s membrane either centrally or peripherally, and focal areas of Descemet’s membrane may be ruptured, similar to that observed in keratoconus.38,39 The areas of Descemet’s membrane rupture may show endothelial repair and scarring of the overlying stroma.40
Keratoglobus may be the result of arrested buphthalmos, a variant of megalocornea, or most likely an extreme form of keratoconus.41
Keratoconus
See the the section on corneal dystrophies.
CONGENITAL OPACITIES OF THE CORNEA
NONSPECIFIC OPACITIES
There are multiple nonspecific types of congenital corneal opacities. Although arrested embryogenesis or intrauterine inflammation may cause the entity, the opacities have the same clinical characteristics as acquired changes following trauma. In order of progressive severity, the degree of opacification is called a facet when only Bowman’s membrane is involved; a nebula when the area of opacification is diffuse, cloudlike, and has indistinct borders; a macula when the area is dense and has a circumscribed border; and a leukoma when the cornea is opaque. Adherent leukoma is a subgroup in which a portion of iris is fused to the posterior surface of the opaque corneal tissue, similar to the findings of some healed acquired corneal perforations (Fig. 2).
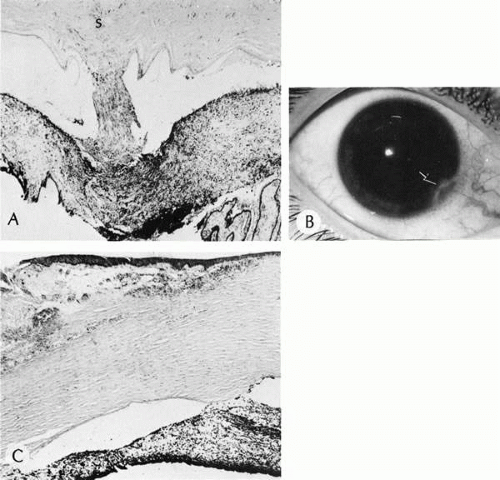 Fig. 2. Adherent leukoma. A. Proliferated fibrous tissue attaches the iris to the cornea through a gap in Descemet’s membrane of a 3-week-old wound. An overlying scar(s) is present through the full thickness of the cornea. After organization and shrinkage of the fibrous membrane, the scar will look much like the scar of adherent leukoma seen in B. Peripheral adherent leukoma (arrow) in a 12-year-old girl who had accidental perforation of the globe by a pair of scissors 5 weeks previously. The perforation of the cornea was repaired on the day of the injury. Sympathetic uveitis developed 2 days before the photograph was taken. C. The peripheral iris is adherent to the corneal stroma through a gap in Descemet’s membrane. The overlying stroma is scarred. (Courtesy of SEI Photoarchives.) |
ANTERIOR EMBRYOTOXON
Anterior embryotoxin, synonymous with arcus juvenilis, consists of a yellow-white linear deposit central and distinct from the limbus and separated by clear cornea (Fig. 3). The condition, seen most often in men, becomes more intense with age. The adult acquired type of deposit, arcus senilis, has been found to be a clinical characteristic of type II hyperlipidemia and is considered to be a risk factor for coronary heart disease and cardiovascular disease.42 The corneal finding is a manifestation of a systemic disease and is not congenital.
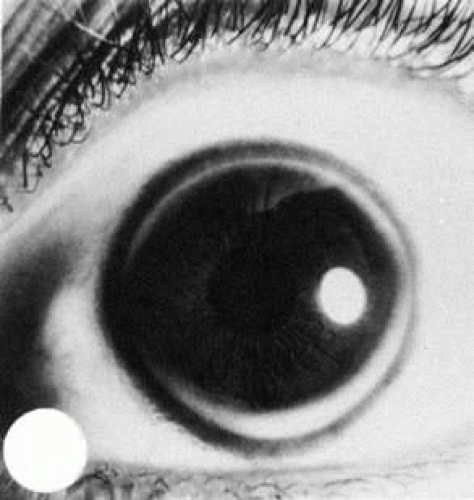 Fig. 3. Anterior embryotoxin (arcus juvenilis) in a 28-year-old woman with familial hypercholesterolemia. |
Lipid can be shown by special staining methods to be located at the junction of the peripheral cornea and sclera, most densely deposited adjacent to Bowman’s membrane, and next to Descemet’s membrane.
The deposit is more likely due to defective clearing of lipid than excessive deposition of lipid.43
CORNEAL KELOIDS
Corneal keloids are hypertrophic scars of the cornea that may be present at birth following intra-uterine trauma. but more often they appear spontaneously or after minor trauma in early childhood. The opacity appears to be an inappropriate repair response of the corneal tissue to trauma. A sector of the cornea or the entire cornea may be involved. The inappropriate repair response usually coexists in the skin. Black persons more commonly are affected than others. Recurrence is the rule, particularly following attempts at surgical excision.44 No inheritance pattern has been recognized.
The stromal nodules are composed of proliferating myofibroblasts, activated fibroblasts, and haphazardly arranged fascicles of collagen. Immunohistochemical stains show spindle cells that express immunoreactivity for vimentin and alpha smooth muscle actin.45
CENTRAL DYSGENESIS OF THE CORNEA
Central dysgenesis of the cornea involves abnormalities of the neural crest mesenchymal derivatives that make up the central cornea posterior to Bowman’s membrane. Bowman’s membrane may be involved secondarily.
PETERS’ ANOMALY
Peters’ anomaly includes absence of central corneal endothelium, Descemet’s membrane, and variable amounts of corneal stroma (Fig. 4). In most cases Bowman’s membrane also is absent. Peters’ anomaly may be caused by primary dysgenesis of the corneal endothelial mesoderm, primary dysgenesis of keratocyte and endothelial neural crest mesoderm, or secondary endothelial degeneration due to late anterior displacement of a normally developed crystalline lens.48 In addition, it has been suggested that abnormal apposition of an ectopic lens to the developing cornea during the second or third month of gestation may be the cause of exceptional cases of peripheral Peters’ anomaly.49
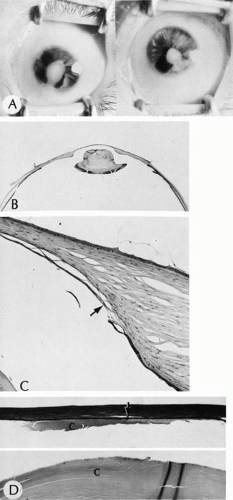 Fig. 4. Peters’ anomaly. A. Note the central corneal scar in the right and left eyes. The lens was adherent to the back of the corneal scar. Iris abnormalities also were present. B. The anterior segment shows a posterior corneal defect, a “top hat” appearance of the lens, and total adherence of the anterior surface of the iris to the cornea. C. High magnification shows termination of the endothelium and Descemet’s membrane (arrow), corneal thinning, and localized absence of Bowman’s membrane. The lens (lower left) is artifactually separated from the cornea. D. A PAS-positive membrane (lens capsule) is shown (top) adherent to the posterior corneal surface (arrow). The lens cortex (c) is artifactually separated from the rest of the lens (bottom). (Courtesy of SEI Photoarchives.) (B–D modified from Scheie HG, Yanoff M: Peter’s anomaly and total posterior coloboma of retinal pigment epithelium and choroid. Arch Ophthalmol 87:525, 1972.) |
Associated anterior segment anomalies include corectopia, iris hypoplasia, anterior polar cataract or other lens abnormalities, and iridocorneal adhesion. (Fig. 5) Corneal perforations secondary to Peters’ anomaly have been reported at birth.50,51 Systemic anomalies include Potter’s syndrome (agenesis of the urinary tract) and intestinal malrotation.52,53,54 Generally, no specific inheritance pattern has been noted, although a family that had an autosomal dominant inheritance pattern has been reported.55
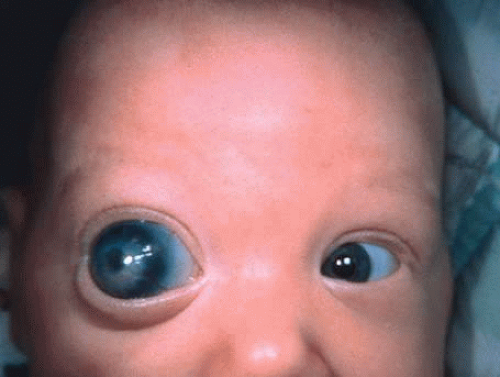 Fig. 5. Peters’ syndrome complicated by buphthalmos. The corneal anterior segment of the right eye has expanded because of the influence of increased intraocular pressure on scleral tissue that is still elastic in young people. |
Histopathologic findings include absence of Descemet’s membrane, corneal endothelium, and usually Bowman’s membrane, as well as thinning of corneal stroma. The defects in Descemet’s membrane, although usually single and central, may be multiple and isolated to the periphery or may be limited to an area of adhesion of iris.56 Descemet’s membrane has been found to have embryonal ultrastructural characteristics combined with attenuated endothelium.57 The corneal stromal lamella are more irregular and closely packed when compared with normal. Immunohistochemical markers indicate that a normal complement of collagens type I, III, IV, V, and VI occurs in Peters’ anomaly; however, an increased concentration may occur of the adhesive protein fibronectin, which is known to play a role in the embryologic development of the cornea.58,59,60
LOCALIZED POSTERIOR KERATOCONUS
Localized posterior keratoconus, which usually presents as an isolated anomaly, consists of central or paracentral depressions of the posterior contour of the cornea. Descemet’s membrane and the corneal endothelium are present. The mechanism of origin is unknown but may be a mild form of Peters’ anomaly. The condition tends to be unilateral, relatively central in the cornea, and sporadic. No relationship exists with anterior keratoconus. Vision is not affected except in extreme cases in which the anterior corneal curvature is secondarily altered. Associated systemic defects include median facial clefting and severe genitourinary abnormalities, which suggests that the defect occurs early in the gestational period.
Histopathologic changes include disarray of corneal stromal collagen in the area of abnormal posterior curvature. Bowman’s membrane has been absent in some cases. In the area of stromal thinning, an abnormal anterior banding and a multilaminar configuration of Descemet’s membrane occurs. Knoblike excrescences of Descemet’s membrane around the periphery of the corneal defect have been observed, suggesting early embryonic iridocorneal adhesion.61,62,63
PERIPHERAL DYSGENESIS OF THE CORNEA AND IRIS (MUTATIONS IN PAX 6 GENE)
The PAX family of genes is highly conserved throughout species indicating the fundamental nature of function in development. The gene products from the PAX genes are necessary for initiating development of certain tissues and organs. The PAX 6 gene is thought to initiate development of the eye. The following group of disparate entities are part of the anterior segment dysgenesis group as illustrated by the Step ladder classification and linked as they involve mutations in at least three genetic loci. The most important reason for segregating these conditions is that they confer a 50% or greater risk of developing glaucoma.64,65
AXENFELD’S ANOMALY
Isolated Axenfeld’s anomaly (posterior embryotoxon) consists of a clinically prominent Schwalbe’s line (terminal end of Descemet’s membrane) plus a variable number of iris processes extending from the peripheral iris to Schwalbe’s line (Fig. 6). The condition is most likely a developmental arrest, late in gestation, of tissues derived from neural crest cells.66 The line appears as a deep linear opacity of the peripheral cornea of variable prominence and extent and is most often found temporally. The prevalence rate is approximately 15% to 25%.67,68 No race or sex predilection exists. Although the majority of the eyes are normal, an associated partial iris coloboma and other anomalies may occur.68 Axenfeld’s anomaly may be associated with non-ocular abnormalities as part of Axenfeld-Rieger’s syndrome (see later).
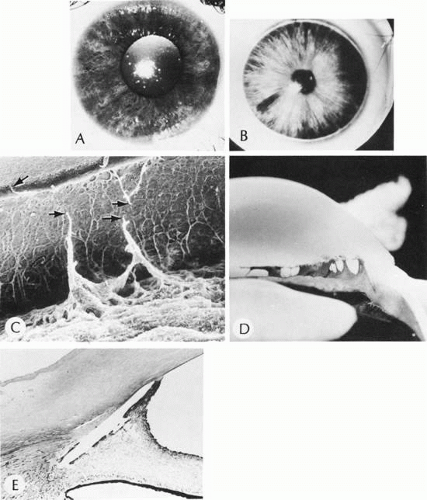 Fig. 6. Axenfeld’s anomaly (posterior embryotoxin). A. The only abnormality visible from the 2-o’clock to 4-o’clock positions adjacent to the limbus is a “ropy” corneal opacity. The other eye is normal. B. A corneal opacity is present over 360 degrees near the limbus at the level of Descemet’s membrane. C. Scanning electron micrograph shows the iris processes spanning the angle and attaching to the anteriorly displaced Schwalbe’s ring. Artifactually broken ends of the iris processes are indicated by the arrows. D. Macroscopic appearance of the iris processes attaching to Schwalbe’s ring. E. Iris processes attach to the anteriorly displaced Schwalbe’s ring. (Courtesy of SEI Photoarchives.) |
Histologically, Axenfeld’s anomaly consists of dense collagen and ground substance covered by a monolayer of flattened endothelial or spindle-shaped cells at the terminal end of Descemet’s membrane. The endothelium is contiguous with the endothelium covering the trabecular beams.68 Associated iris processes are composed of normal-appearing iris stroma.
RIEGER’S SYNDROME
Peripheral dysgenesis (isolated Rieger’s syndrome) encompasses a wide spectrum of developmental abnormalities of anterior chamber angle tissues of neural crest origin associated with systemic anomalies.70 This group of abnormalities is important clinically because it is associated with an increased prevalence of glaucoma. The defects are thought to result from a developmental arrest in the third month of gestation. Rieger’s syndrome probably includes those entities sometimes described as mesodermal dysgenesis and anterior chamber cleavage syndrome and most accurately called anterior segment dysgenesis.
Histopathologic changes are characterized by retention of primordial endothelial tissue on the iris and by anterior chamber angle and peripheral iris strands (Fig. 7). Continued contraction of component membranes causes progressive changes of the iris architecture. Glaucoma results from arrested development of the anterior chamber angle structures, characterized by incomplete maturation of the trabecular meshwork and Schlemm’s canal and a high insertion of the peripheral iris.70
 Fig. 7. Rieger’s syndrome. A. Posterior embryotoxon, marked iridocorneal processes and iris hypoplasia are present. B and C. Arrows show the central location of Schwalbe’s ring. (Courtesy of SEI Photoarchives.) |
AXENFELD-REIGER SYNDROME
Because Rieger’s syndrome is often associated with Axenfeld’s anomaly as one of its ocular findings, along with marked anomalous development of the iris and systemic anomalies mainly consisting of facial and dental abnormalities, the clinical term Axenfeld-Rieger syndrome has been suggested.66 Corectopia (displacement in the direction of prominent peripheral tissue strands), dyscoria, slit pupil, iris hypoplasia, and prominent iris strands have been reported. Glaucoma eventually may be found in 50% of affected people; however, the glaucoma may not be manifested until childhood or early adulthood. Extraocular abnormalities include enamel hypoplasia, conical and misshapen teeth, hypodontia; impactions, underdevelopment of the maxilla, mandible, and cranial base (anterior and posterior), low-set ears, wide nasal bridge, bilateral microcondyles, and bilateral choanal atresia. Autosomal dominant inheritance patterns in families have been identified.72
Three chromosomal loci have recently been demonstrated to link Axenfeld-Rieger syndrome and related phenotypes. These loci are on chromosomes 4q25, 6p25, and 13q14. The genes at chromosomes 4q25 and 6p25 have been identified as PITX2 and FKHL7, respectively. Mutations in these genes can cause a wide variety of phenotypes that share features with Axenfeld-Rieger syndrome. Axenfeld anomaly, Rieger anomaly, Rieger syndrome, iridogoniodysgenesis anomaly, iridogoniodysgenesis syndrome, iris hypoplasia, and familial glaucoma iridogoniodysplasia all have sufficient genotypic and phenotypic overlap such that they should be considered one condition.64
DYSGENESIS OF THE CORNEA
CONGENITAL CORNEAL ECTASIA
Congenital corneal ectasia is an opaque, ectatic cornea extending between the lids. If the ectactic cornea is lined by adherent iris, the condition is called corneal staphyloma. The cornea then has a blue hue caused by posterior approximation of atrophic iris tissue. Lens opacities are common. The posterior segment is normal. No inheritance pattern is evident. Congenital corneal ectasia is thought to be due to failure of migration of embryonic mesoderm to form corneal endothelium and iris stroma at approximately 7 weeks’ gestation.73,74
Histologically, the corneal epithelium is normal in thickness but may be keratinized secondary to exposure. Often, local attenuation of Bowman’s membrane occurs. The stroma is thickened, disorganized, hypercellular, and vascularized. A double layer of pigment-containing cells lines the posterior corneal stroma. Usually no sign of an inflammatory infiltrate is present. Descemet’s membrane and corneal endothelium are absent.
Corneal ectasia has become a major concern related to laser-assisted in-situ keratomileusis (LASIK) surgery. Increasing numbers of cases of progressive corneal ectasias have been reported, with many leading to keratoconus.75
SCLEROCORNEA
Sclerocornea is a totally opacified cornea that shows clinical and histologic features of sclera. The condition often is bilateral. Superficial or deep vascularization of the tissue may occur. Associated clinical conditions include nystagmus, strabismus, aniridia, cornea plana, macular hypoplasia, horizontally oval cornea, and glaucoma.76 As described in Mieten’s syndrome, sclerocornea may present with congenital cerebral dysfunction, deafness, cryptorchidism, pulmonary disease, brachycephaly, and defects of the face, ears, and skin. Sclerocornea may be inherited in an autosomal dominant pattern. It may result from intrauterine inflammation and other nonspecific causes.
Histologically, increased numbers of collagen fibrils with a variable collagen diameter occur in the normal corneal stroma. Descemet’s membrane appears thin.77,78 Bowman’s membrane may be absent.79
CHORISTOMAS OF THE CORNEA
CORNEAL DERMOID
Corneal dermoids are choristomas of mesenchymal elements covered by epithelium. The location and extent are variable. Corneal dermoids usually are sporadic; however, autosomal recessive or sex-linked pedigrees have been described.83
The most common location is at the limbus, where the abnormal tissue is present in the peripheral cornea and in the adjacent episclera. Although the majority of limbal dermoids are isolated findings, approximately 30% are associated with Goldenhar’s syndrome (see later). Although most limbal dermoids are superficial, the abnormal tissue occasionally extends into the anterior chamber angle tissue. Central corneal dermoids are the least common (Fig. 8). The posterior cornea and Descemet’s membrane are normal. On rare occasions the entire cornea and anterior segment may be replaced by a dermoid. Descemet’s membrane, iris, anterior chamber, and crystalline lens may be absent. This severe type often is associated with microphthalmos.84
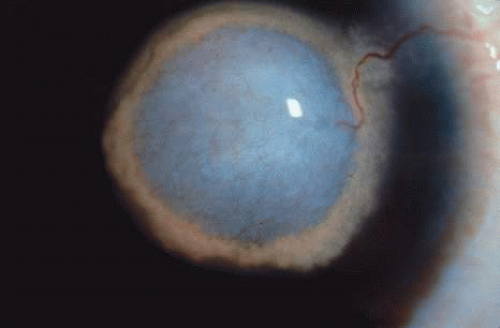 Fig. 8. Central corneal dermoid. The entire cornea is opaque and vascularized. |
Histologically, the corneal epithelium may be keratinized. Bowman’s membrane often is absent. The stroma is replaced to a variable degree by irregularly arranged, dense, vascularized, collagenous connective tissue containing hair follicles, hair shafts, sebaceous glands, fat, smooth muscle, striated muscle, cartilage, teeth, or bone. The mass may be either cystic or solid.
GOLDENHAR’S SYNDROME
Goldenhar’s syndrome consists of bilateral epibulbar dermoids, accessory auricular appendages, blind pretragal fistulas, and abnormalities of the cervical vertebrae85 (Fig. 9). The condition is important because of associated systemic abnormalities, including mandibulofacial dysostosis, phocomelia, and renal malformations. The first and second brachial clefts give rise to the ear, face, and eyelids. Goldenhar’s syndrome is thought to result from a noxious insult during the seventh week of gestation, affecting the brachial clefts and a variable number of other germ tissues of other organ systems, primarily the kidney and skeleton. Associated brachial arch alterations include notching (so-called coloboma) of the upper eyelid, antimongoloid slant of the palpebral fissures, microphthalmos, microcornea, uveal coloboma, hypoplastic upper and lower jaw, microtia, and macrostomia. Nonbrachial arch alterations include anomalies of the cervical vertebrae, heart disease, cleft palate, mental retardation, hydrocephalus, meningioencephalocele, phocomelia, renal hypoplasia, and partially annular pancreas.86
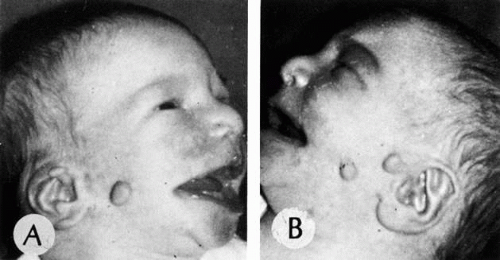 Fig. 9. Goldenhar’s syndrome. A and B. Accessory auricular appendages and aural fistulas are present. |
Although the eye usually is not involved, a subgroup identified as the Goldenhar-Gorlin syndrome shows diminished visual acuity, tilted optic disc, optic nerve hypoplasia, tortuous retinal vessels, macular hypoplasia and heterotopia, microphthalmia, and anophthalmia. The Goldenhar-Gorlin syndrome may be caused by an asynchrony in the migration of the neural crest cells in the early stages of embryonal development.87
INFLAMMATORY CONDITIONS OF THE CORNEA
Inflammatory conditions of the cornea are difficult to classify. For example, herpes simplex can present in a nonulcerative or ulcerative form. The viral infection and associated inflammation can involve the epithelium, stroma, and endothelium either concurrently or sequentially. With this proviso in mind, the various forms of inflammation are presented here in terms of nonulcerative, epithelial, stromal, and endothelial clinical presentations. Ulcerative keratopathy is discussed separately, although the condition may not be ulcerative when first diagnosed.
NONULCERATIVE EPITHELIAL INFLAMMATION
Epithelial inflammation often is a part of primary conjunctival inflammation (e.g., almost any form of severe bacterial conjunctivitis). Epithelial inflammatory changes in the form of superficial epithelial punctate keratitis or recurrent erosion syndrome may be secondary to abnormalities of underlying tissue, such as inherited corneal dystrophies (e.g., Reis-Bucklers’ dystrophy and lattice, granular, and macular dystrophies).
Histologically, polymorphonuclear leukocytes are found insinuating among the squamous epithelial cells (exocytosis) associated with intercellular edema (spongiosis) and intracellular edema (primarily of the basalar cell layer). The basement membrane of the epithelium, which usually is inconspicuous by light microscopy, may become very prominent. The orderly maturation of the squamous epithelium may be disturbed, causing various degrees of cellular atypia. In severe and prolonged cases, serous-filled subepithelial bullae may develop. If the integrity of the superficial epithelium is disrupted (recurrent corneal erosion), the anterior corneal stroma may become inflamed and subsequently scarred. With resolution of the edema and bullae, the cells may become maloriented relative to Bowman’s membrane, causing intraepithelial basement membrane formation and intraepithelial cyst formation (epithelial basement membrane syndrome).
THYGESON’S SUPERFICIAL PUNCTATE KERATITIS
Thygeson’s disease is a primary inflammation of the cornea of unknown cause that usually occurs in young people, often between the ages of 20 and 40 years. The patients present with severe symptoms: tearing, foreign body sensation, and photophobia. The initial clinical sign is an outcropping of fine, ground glass–like particles arising from the superficial epithelial layer that mass together as coarse clumps 0.5 to 1 mm in diameter. The lesions usually remain for several weeks to several months and eventually fade, only to recur months or years later. Thygeson’s disease is one of the few corneal epithelial inflammatory conditions that present with an elevation of the epithelial surface, which stains with fluorescein in a negative pattern (i.e., the fluorescein pools around the base of the elevation and the apex stains negatively).
By light microscopy the epithelial cells show a cytopathic effect in the acute stages, suggestive of a viral infection; however, no viral particles have been identified by electron microscopy. Only rarely have viral cultures been positive, and most of these isolated reports have not been confirmed.88,89
SUPERIOR LIMBIC KERATOCONJUNCTIVITIS OF THEODORE
Superior limbic keratoconjunctivitis of Theodore is an idiopathic inflammation of the upper tarsus, superior limbus, and corneal epithelium associated with limbal follicles and the development of recalcitrant filamentary keratitis in one third of cases. Keratoconjunctivitis sicca is a frequent associated finding.90 The patients present with fine, papillary, upper tarsal conjunctival reaction in all cases. The superior bulbar conjunctiva often shows positive fluorescein and rose bengal staining. An association with thyroid disease exists in almost 50% of affected individuals.
The corneal alterations are secondary to irritation by the superior tarsal papillary reaction, which results in the disturbance of the superior bulbar conjunctiva, leading to the development of tenacious mucus, which in turn leads to filamentary keratitis. If not treated or in severe cases, the superior bulbar conjunctiva may undergo epidermalization (transformation of mucous membrane to a tissue with the characteristics of skin, primarily keratinization).
Histologic changes of the conjunctiva consist of hyperplasia of the squamous layer and induction of a granular layer that indicates keratin production. The presence of keratinized epithelium either by superficial scraping or full-thickness biopsy supports the clinical diagnosis but is not specific.91
EPIDEMIC KERATOCONJUNCTIVITIS
Epidemic keratoconjunctivitis (EKC) is a highly contagious, self-limited subepithelial inflammatory disease associated most often with adenovirus type 8 and less commonly with adenovirus types 19 and 25.92 The disease often occurs as epidemics in factories, schools, or offices of ophthalmologists. The viral particles remain infectious for up to 14 days on metal, glass, or plastic surfaces (e.g., tonometer heads). An incubation period of approximately 8 days occurs between infection and clinical expression in the first eye followed 3 to 7 days later by expression in the second eye. The second eye usually is involved less severely. The onset often is abrupt or explosive, consisting of follicular conjunctivitis associated with tearing, marked swelling and hyperemia of the conjunctiva, and ipsilateral, painful preauricular lymphadenopathy (Fig. 10). In children, EKC can mimic the presentation of orbital cellulitis.93 Conjunctival membranes may develop during the course of EKC, which in turn may lead to filamentary keratitis and/or frank symblepharon formation. Subepithelial keratitis occurs during the second or third week, often reducing visual acuity significantly (20/60).94
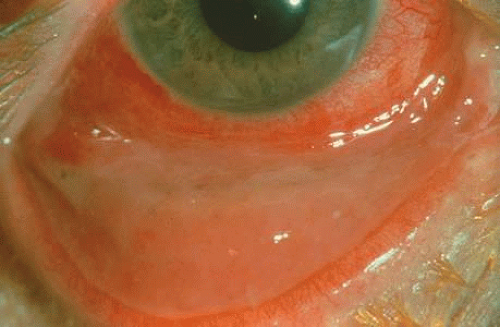 Fig. 10. Inflammatory membrane associated with epidemic keratoconjunctivitis (EKC) caused by adenovirus type 8. The entire inferior tarsal conjunctive is covered by thick, tenaceous, fibrinous exudate. |
Historically, very few tissue specimens have been studied. Initially, lymphocytes infiltrate the subepithelial area of the cornea, followed by mild scarring. The resulting nebula may be permanent.
TRACHOMA
Trachoma is a chronic follicular conjunctivitis caused by a unique class of organisms, Chlamydiaceae, characterized by small size, a gram-negative staining pattern, a biphasic life cycle, and sensitivity to antibiotics.95 The metabolically inert, extracellular form of the organism (elementary body) infects specific epithelial cells of the body. Several of the serotypes of Chlamydia trachomatis (A, B, Ba, C) infect only the epithelial cell of the conjunctiva. Other serotypes are known to infect epithelial cells in other parts of the body, causing other specific disease entities (e.g., lymphogranuloma venereum and psittacosis). Once inside the cell, the organism transforms to the metabolically active obligate intracellular parasite (reticulate body), which ultimately forms a microcolony of new elementary bodies. This intracytoplasmic inclusion is typically perinuclear and has been called the Halberstaedter-Prowazek inclusion. The elementary bodies are released at the death of the host cell and infect other epithelial cells.
Corneal blindness caused by this disease has been described in the earliest medical texts and has influenced the history of Europe, North Africa, the Middle East, and Asia. The organism only infects humans. It is spread from person to person, including mother to child, by direct contact with mucous secretions or by the use of common towels or eye makeup. The people most at risk are those living in close quarters, often associated with poor economic, nutritional, and social status. The acute form of trachoma presents in children as the lymphoid tissue of the conjunctiva becomes mature. Extensive mucoid discharge, conjunctival hyperemia, and a myriad of follicles, primarily on the upper tarsal plate, are the initial manifestations. The clinical signs may wax and wane, depending partly on the frequency and severity of secondary bacterial infections. Following a variable period of recurrent episodes of acute or subacute conjunctivitis, the inflammatory phase is replaced by cicatricial changes of the upper tarsal plate, leading to severe trichiasis and entropion. The mechanical trauma from the scarred lid margin on the cornea causes extensive superficial scarring of the cornea beginning with a delicate superior vascular pannus and culminating in a densely opaque, scarred cornea.
Histologically, desquamated epithelial cells or cells sampled by tarsal conjunctival scraping contain discrete, round, densely staining spheroids (initial bodies) that aggregate into a well-defined, paranuclear intracytoplasmic inclusion called the elementary body (Halberstaedter-Prowazek). A distinct rim of cytoplasm between the elementary body and the adjacent nuclear membrane distinguishes the infective agent from crush artifact of the nucleus. The epithelial remnants of cells that have been lysed by the infection often are seen in large phagocytic cells (Leber cells). In the early phases of the disease, the conjunctival subepithelial tissue is markedly expanded by reactive lymphoid hyperplasia, characterized by numerous germinal centers in a polymorphic lymphoid infiltrate. Follicles may form in the paralimbal tissue and ultimately involute to form Herbert’s pits. Corneal neovascularization in the form of an inflammatory pannus begins in the superior cornea in the region of the limbal inflammatory infiltrate.96 Associated secondary bacterial conjunctivitis may coexist, as evidenced by an acute, sometimes suppurative, inflammatory reaction. With time, the inflammatory reaction is superseded by an extensive subepithelial tarsal conjunctival fibrovascular reaction, laying down collagen and contracting in a plane just above and parallel to the lid margin (Arlt’s line). The mechanical distortion of the tarsal plate and concurrent destruction of the tarsal plate integrity are noted clinically as entropion and trichiasis. Goblet cells in the conjunctiva are destroyed by the prolonged inflammatory reaction. Subepithelial scarring compromises accessory lacrimal gland tissue and the ducts of the main lacrimal gland. Subsequent anterior surface drying accelerates the scarring process. The scarring of the cornea is a nonspecific degenerative and inflammatory pannus initially, ultimately resulting in the total loss of Bowman’s membrane and scarring of the superficial stroma. The corneal tissue eventually may be substantially replaced by secondary amyloidosis.97
LEPROSY
Leprosy (Hansen’s disease) is an infectious disease caused by Mycobacterium leprae, a gram-positive, obligate intracellular bacillus of human beings. The disease is characterized by slowly progressive chronic inflammation of the skin, nerves, and eyes. When cell-mediated immunity is maintained (tuberculoid leprosy), the disease is manifested by a small number of cutaneous lesions associated with marked nerve anesthesia and enlargement. Few organisms are seen in histologic sections from tuberculoid leprosy lesions. In individuals who have compromised cell-mediated immunity (lepromatous leprosy), numerous skin lesions occur, associated with less anesthesia. Many organisms are present in mononuclear or epithelioid cells (lepra cells). The lepromatous type is the most contagious. The organism has a particular affinity for nerves and for cooler areas of the body. The cornea, iris, and ciliary body generally are several degrees cooler than core body temperature and are therefore vulnerable to infection.98
The anterior segment may be directly infected by M. leprae through the blood stream (chronic anterior uveitis, enlargement of corneal nerves). Inflammation of the facial nerve may lead to exposure keratopathy because of neurogenic lid dysfunction. Immune-complex disturbance may cause episcleritis, scleritis, and iridocyclitis. Impaired corneal sensation disrupts normal corneal metabolism due to lack of substance P and repeated unrecognized trauma. Mechanical abnormalities of the lids may cause direct corneal trauma.99
The histologic changes in the cornea are influenced by the immune competence of the host. In the tuberculoid form, the cornea is infiltrated with chronic granulomatous inflammation and minimal scarring unless the cornea has been secondarily infected or severely traumatized. In the lepromatous form, the epithelioid cells of the inflammatory infiltrate are filled with viable organisms, giving the cytoplasm a granular appearance.
ROSACEA
Rosacea keratitis is the corneal component of acne rosacea (Fig. 11). The cause of acne rosacea is undetermined but is thought to be a genetically inherited abnormal vasodilation response of the skin, associated with sebaceous hyperplasia and a chronic inflammatory reaction. Approximately 3% of patients with acne rosacea have involvement of the cornea, whereas 20% of patients have eyelid and conjunctival involvement.100 Corneal changes include peripheral subepithelial scarring, which progresses to the axial cornea, accompanied by stromal vascularization.
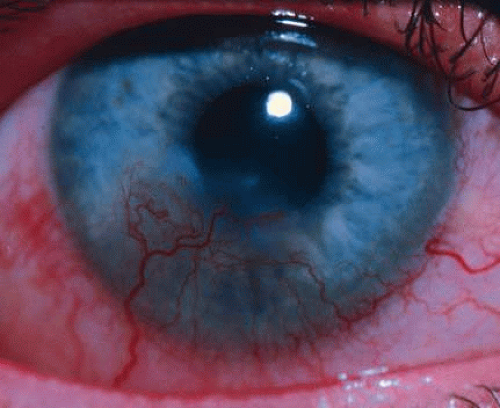 Fig. 11. Rosacea keratitis with characteristic inferior vascularized pannus encroaching upon the visual axis. |
Early in the disease, a superficial lymphocytic infiltrate is found in the subepithelial-superficial stromal corneal tissues, leading to sclerosing pannus formation. The process characteristically begins in the inferior peripheral cornea and progresses to the axial cornea.
STROMAL OR “INTERSTITIAL” KERATITIS
Syphilis
Syphilis is a venereal disease caused by the spirochete Treponema pallidum, which primarily affects the central nervous and cardiovascular systems. During the past decade, syphilis has again become more common. Often syphilis is found in patients who also have acquired immune deficiency syndrome (AIDS).101 The organism is highly infectious but of low virulence, resulting in long periods of latency and prolonged viability unless specifically treated. Many of the tissue effects of syphilis are due to host immune response, such as mononuclear cell infiltrates, proliferative vascular changes, and occasionally granuloma formation. The cornea often is not affected by acquired syphilis but is commonly affected by congenital syphilis (Fig. 12). Infection of the fetus occurs transplacentally after the fifth month of gestation. Diffuse fibrosis can compromise the function of any parenchymatous organ, including the lungs.
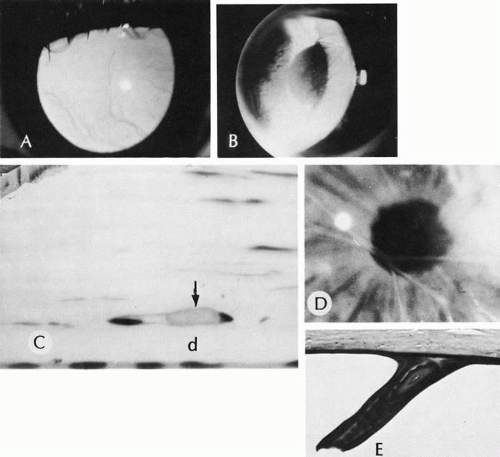 Fig. 12. Syphilis. A. Corneal ghost vessels as viewed by fundus reflex in a patient with congenital syphilis. B. Slit lamp appearance of interstitial keratitis. C. A blood vessel (arrow) present anterior to Descemet’s membrane (d). D. Retrocorneal ridges of Descemet’s membrane form refractile, branching straight lines. E. A multilayered strand extends from a thickened Descemet’s membrane into the anterior chamber.(Courtesy of SEI Photoarchives.) (B Courtesy of Dr. W. C. Prayer; D and E from Waring GO, Font RL, Rodrigues MM et al: Alterations of Descemet’s membrane in interstitial keratitis. Am J Ophthalmol 81:773, 1976.) |
The cornea is particularly involved in a late-occurring form of congenital syphilis, which also causes periostitis, saber chins, saddle nose deformity, and tooth deformities (Hutchinson’s teeth).102 Congenital syphilis presents between the ages of 5 and 10 years with an intense keratitis that may last for several months and may reduce visual acuity to counting fingers or seeing hand movements.103 Fortunately, usually a significant regression occurs with a parallel improvement of visual acuity, often in the range of 20/40 to 20/60. The acquired form of interstitial keratitis tends to be unilateral (it may even be sectorial) and tends to occur during the third or fourth decade of life as an expression of tertiary syphilis.
Histologically, the cornea shows edema and infiltration by lymphocytes and plasma cells. Vessels usually are seen in the deep portion of the cornea, just anterior to Descemet’s membrane. Although the edema and inflammation of the corneal stroma resolves, the deep vessels persist in the form of ghost vessels. Often blood flow is minimal but persistent through the vessels, even though they appear empty. Chronic interstitial inflammation causes alterations of Descemet’s membrane that are characteristic of congenital syphilis and include linear guttae with ridges and even nests of transparent basement membrane material, which may project into the anterior chamber.104,105
The association with uveitis is frequent and occasionally may lead to significant synechia formation. It should be noted that in the anterior chamber, antigens to T. pallidum may be found.
LYME DISEASE
Lyme disease, named for the Connecticut town in which it was first recognized, is the result of systemic infection by the spirochete Borrelia burgdorferi.106 There are three stages: primary (erythema chronica migrans)—skin inoculation through bites by the nymphs and hard ticks of the genus Ixodes; secondary—systemic dissemination of the spirochete; and tertiary—involvement of the joints, central nervous system, and cardiovascular system.
Ocular findings are relatively uncommon and include nummular keratitis, uveitis, retinitis, and optic neuritis. It appears that all forms of ocular involvement that have occurred as a manifestation of syphilis also can be caused by B. burgdorferi. However, the organism has never been found inside the eye.107
TUBERCULOSIS
Tuberculosis is a granulomatous disease caused by Mycobacterium tuberculosis, which primarily affects the lungs and kidneys. Tuberculosis of the cornea occasionally presents as an extension from conjunctival disease as a superior pannus. Rarely, it may present as an interstitial keratitis. The organism is not found in the cornea. On the other hand, M. chelonae is often found in the cornea, as are several other atypical mycobacteria.108
SARCOIDOSIS
Sarcoidosis is a generalized granulomatous inflammation of unknown cause primarily involving the lungs. Sarcoidosis may affect the conjunctiva but rarely affects the cornea directly. Occasional cases occur as an extension from the conjunctiva or uvea.109
ONCHOCERCIASIS
Onchocerciasis is an infectious disease caused by the largest human filarial worm, Onchocerca volvulus. The microfilaria of the worm is transmitted to human subcutaneous tissues by the bite of the blackfly Simulium damnosum. The microfilaria matures in the skin into an adult worm, which may be up to 50 cm in length. The microfilaria discharged from the adult worm migrates through the interstitium of the skin rather than hematogenously to the eye. Alive, the microfilaria are fairly well tolerated and induce only a slight, surrounding lymphocytic or plasma cell reaction. Dead organisms, however, cause a severe inflammatory reaction that is chiefly eosinophilic. The inflammation may cause corneal opacification directly or indirectly by causing secondary iritis with synechia formation and angle closure glaucoma.110
VIRAL KERATITIS
Herpes Simplex Stromal Keratitis
Herpes simplex stromal keratitis is stromal inflammation occurring with or after epithelial infection by herpes simplex virus. Occasionally, disciform keratitis may be the initial presentation of the infection (Fig. 13). Recently it has been found that some strains of herpetic virus may be associated with disciform keratitis.111 Viral cultures of stroma or epithelium or both tend to be negative. However, herpes simplex antigens have been identified in the corneal stroma of 50% of the cases studied.112 The presence of viral antigens suggests that the stromal inflammation is due to host reaction directed against the antigens or to viral particles.
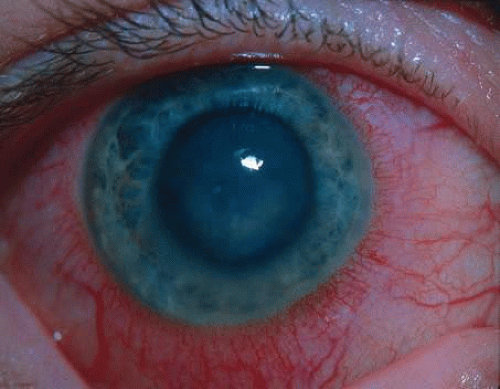 Fig. 13. Herpes simplex keratitis can form a dense stromal opacity, ususally after multiple episodes of epithelial infection. However, a patient may also present with this finding associated with underlying large keratitic precipitates. |
Histologically, edema separating corneal lamellae, associated with an infiltrate of polymorphonuclear leukocytes, is present early, followed promptly by an infiltrate of lymphocytes and plasma cells. Characteristically, a granulomatous reaction to Descemet’s membrane occurs; the membrane may harbor viral particles.113 The inflammation causes destruction of the corneal stromal lamellae, which ultimately may cause perforation.
Herpes Zoster Keratitis
Herpes zoster keratitis is superficial keratitis usually occurring in more severe cases of herpes zoster ophthalmicus, associated invariably with significant scleritis and uveitis. Corneal involvement is caused by ischemia associated with inflammatory occlusion of the ciliary vasculature and anesthesia associated with perineural inflammation of the ciliary nerves.114 Viral particles have not been identified in the cornea. Late in the disease, neurotrophic keratitis can occur, which may lead to exposure keratopathy and corneal perforation secondary to ulceration.
The corneal tissue may be infiltrated with lymphocytes associated with nonspecific degenerative changes. Rarely, a prominent polymorphonuclear leukocyte infiltrate occurs in patients who develop anterior segment necrosis. Corneal perforation is relatively uncommon. The affected corneal lamellae are frequently replaced by vascularized scars that often leak lipid into the cornea, leading to progressive visual loss. Characteristically, a granulomatous reaction to Descemet’s membrane occurs, as seen in herpes simplex and fungal keratitis.
COGAN’S SYNDROME
Cogan’s syndrome consists of nonsyphilitic interstitial keratitis and characteristically patchy, bilateral vascularization of the middle and deep corneal stroma, associated with vestibular auditory symptoms, such as hearing loss, dysacousia, and vertigo. The cause of the syndrome is not known. However, there appears to be some form of systemic vasculitis with inflammation found in the dura, gastrointestinal tract, spleen, and kidneys.115 There is no association with immune abnormalities or specific HLA antigen patterns. This disease most frequently is found in young people; however, it can occur in older age groups. The corneal disease tends to be relatively mild, although vascularization can be a significant problem. Approximately 10% of patients exhibit an underlying vasculitis, primarily involving larger vessels (proximal aortitis, aortic insufficiency).116
OTHER SYSTEMIC DISEASES
Other diseases that can cause secondary stromal keratitis are Hodgkin’s disease, lymphogranuloma venereum, hypoparathyroidism, mycosis fungoides, and gold toxicity.
INFLAMMATION—ULCERATIVE
Peripheral Ulceration
Marginal corneal ulcers usually occur in the interpalpebral region just within the limbus, and they tend to be solitary. Most of these lesions are secondary to allergic reactions to bacterial toxins (e.g., Staphylococcus) or due to direct effects of bacterial proteins. Herpes simplex virus infection also may present as a secondary peripheral corneal ulceration.
The characteristic marginal ulcer secondary to allergic reactions to bacterial toxins presents as an infiltrate composed mainly of small lymphocytes in the superficial corneal lamellae.
Phlyctenular Disease
A phlyctenular ulcer is an inflammatory response associated with staphylococcal toxins, tuberculosis keratitis, or acne rosacea or without known case.117 The lesions occur in children and appear as a small, pinkish-white elevations in the cornea adjacent to the limbus. Ultimately the elevations develop a central gray crater.
Ring Ulcer
A ring ulcer is a coalescence of multiple marginal ulcers found usually in patients who have severe systemic disease or who are debilitated. The individual ulcers begin in the form of superficial marginal keratitis just inside the limbus.
Histologically, the cornea shows involvement with polymorphonuclear leukocytes in the area of necrosis and with lymphocytes and plasma cells in the adjacent tissue. An occlusive vasculitis may be present (Fig. 14).
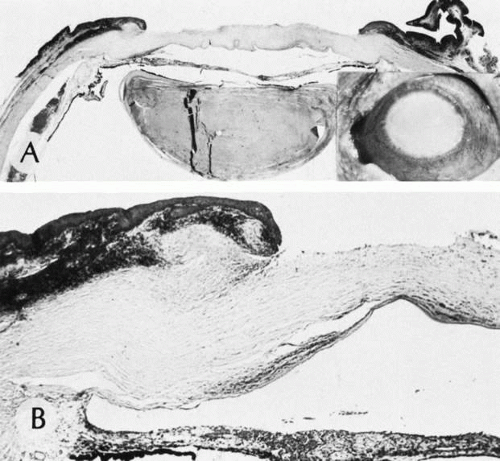 Fig. 14. Ring Ulcer. A. Inset shows a ring ulcer in a debilitated patient, which has spread to involve most of the cornea. B. High magnification shows the peripheral edge of a corneal ulcer.(Courtesy of SEI Photoarchives.) |
Ring Abscess
A ring abscess often is confused with ring ulcer; a ring abscess usually is secondary to serious intraocular disease (e.g., bacterial or fungal endophthalmitis). Occasionally, a ring abscess may be the presenting sign of collagen vascular disease. In either case, a ring abscess is a prognostic sign of poor visual outcome. Histologically, the cornea is infiltrated with polymorphonuclear leukocytes and contains necrotic stromal lamellae and the remnants of degranulated leukocytes.
CENTRAL CORNEAL ULCERS
Bacterial Corneal Ulcers
The specific bacteria causing a central corneal ulcer usually cannot be determined by the clinical appearance of the ulcer, although certain clinical clues may be evident; Pseudomonas tends to cause more rapid liquefactive necrosis than does Pneumococcus (Fig. 15). However, Gram staining of the ulcerated tissue and associated necrotic debris can determine the type of bacteria in 50% of cases.118 The specific bacterial agent responsible for ulceration can be determined the majority of the time by culture.119 The most commonly found organisms include P. aeruginosa, Streptococcus pneumoniae, and Staphylococcus aureus. Other organisms can be found at a much lower frequency.
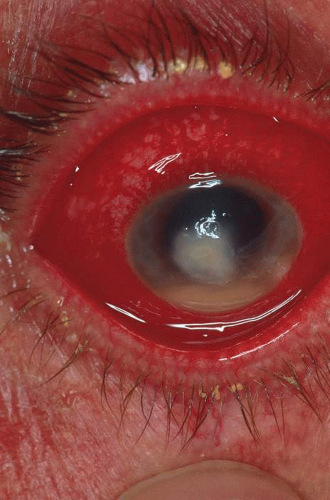 Fig. 15. Pseudomonas keratitis in an 18-year-old otherwise healthy contact lens wearer. |
Initially, there is an infiltrate of acute inflammatory cells in the anterior corneal stromal lamellae, which accumulates in the central cornea. The overlying epithelium and corneal stroma ulcerate. Frank colonies of bacteria may be found in the anterior corneal stroma and within the necrotic debris of the ulcer crater. The corneal inflammation usually is associated with a sizable hypopyon (Fig. 16). The hypopyon always is sterile because bacteria cannot pass through an intact Descemet’s membrane.
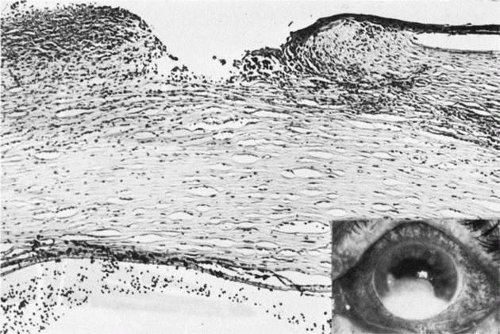 Fig. 16. Central corneal ulcer filled with necrotic debris. Note the neutrophils in the anterior chamber (hypopyon) and infiltrating into the cornea. Corneal ulcer (outlined by fluorescein) and a large hypopyon are seen (inset). (Courtesy of SEI Photoarchives.) |
Viral Corneal Ulcers
HERPES SIMPLEX CORNEAL ULCERS.
Herpes simplex is the most common cause of viral central corneal ulcers in the United States.120 The initial clinical sign may be a cutaneous vesicular rash (Fig. 17) or a corneal epithelial dendrite (Fig. 18). The infection progresses through corneal stromal inflammation (metaherpetic phase) to culminate in central ulceration. The initial epithelial defect is caused by replication of herpes simplex virions in the epithelial cells; the cytopathologic effect of the infection leads to epithelial cell death (rose bengal–positive) and ulceration. The infection is self-limited to an 8- to 10-day course unless shortened by debridement or antiviral therapy. The recurrence rate of initial dendritic infection by herpes simplex (25%) is not influenced by the type of treatment used. Most of the significant complications, such as corneal ulceration (Fig. 19), of herpetic keratitis are related to its tendency to initiate a delayed hypersensitivity type IV reaction, rather than the result of direct infection of the stroma.
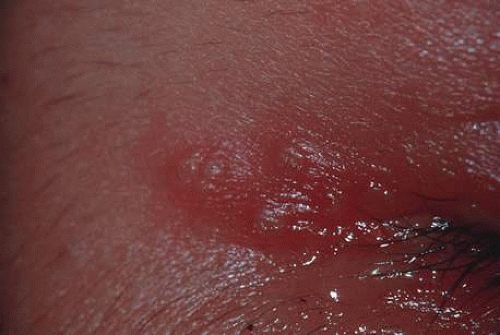 Fig. 17. Herpes simplex keratitis may present as a vesicular eruption of the skin prior to any direct corneal involvement. |
 Fig. 18. Herpes simplex keratitis most often presents with a cytopathologic epithelial change from viral infection of contiguous cells. The infected cells are often linked in a branching or dendritic distribution. Topical fluorescein staining or rose bengal staining highlights the infected cells. |
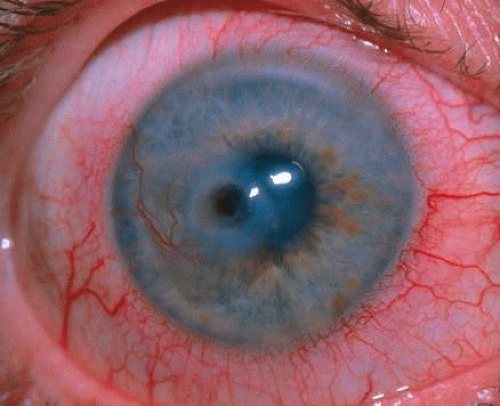 Fig. 19. Ulceration of herpetic keratitis resulting in descemetocele formation. |
By light microscopy, eosinophilic intranuclear inclusions are characteristic of herpes simplex viral infections and represent the result of viral replication (Cowdri type A inclusions).121 Hypersensitivity type IV reactions in the stroma are characterized by lymphocytic and plasmacytic infiltrates. Intranuclear inclusions are relatively rare and are not found in most specimens. Viral particles occasionally can be found in multinucleated giant cells or within the stroma, especially in keratocytes (Figs. 20 and 21).
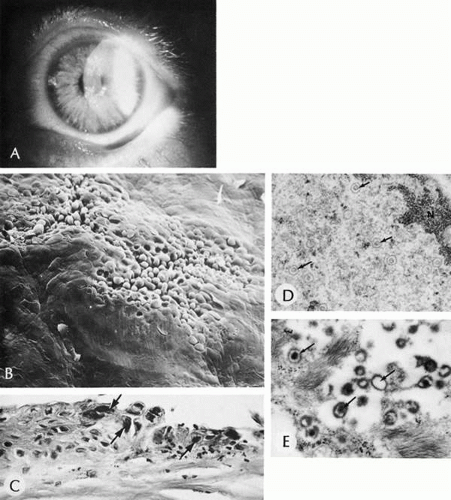 Fig. 20. Herpes simplex. A. Typical dendritic ulcer. B. Scanning electron micrograph of a dendritic ulcer in the epithelium of a rabbit cornea. C. Many intranuclear inclusions (arrows) are present in the corneal epithelium near the edge of the ulcer. D. Virus particles (arrows) of herpes simplex are present in the nucleus. E. Virus particles also are present within the cytoplasm. Note the large size of the cytoplasmic virions. Some particles show empty capsids, whereas others are complete, containing nucleoids. (Courtesy of SEI Photoarchives.) (B Courtesy of Dr. R. C. Eagle Jr; C from Font RL: Chronic ulcerative keratitis caused by herpes simplex virus. Arch Ophthalmol 90:382, 1973.) |
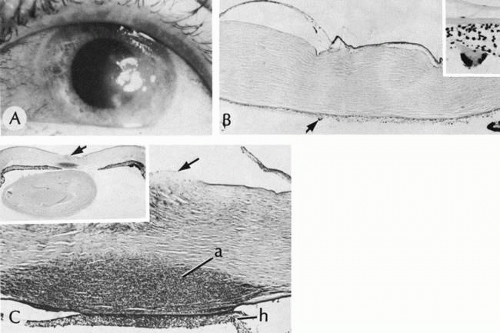 Fig. 21. Herpes simplex. A. Clinical appearance of bullous keratopathy. B. Chronic condition shows development of bullous keratopathy. The anterior chamber inflammatory reaction contains multinucleated inflammatory giant cells (arrow), shown under high magnification in inset. C. Ulcerated bullous keratopathy (arrows). A corneal abscess (a) and hypopyon (h) are present. Note (inset) the subluxation of the lens to left, caused by the loss of zonula-lens attachments on the right, resulting in a “blunted” appearance of the right side of the lens. (Courtesy of SEI Photoarchives.) |
Other Viral Corneal Ulcers
Other viral-induced, central cornea ulcerations are uncommon with the exception of herpes zoster. The ulcer of herpes zoster most often is the result of exposure rather than viral infection or hypersensitivity.
Fungal Corneal Ulcers
Three fungal organisms are responsible for 80% of mycotic keratitis. The most common organism is different in different geographic regions of the United States: Candida albicans in the north and northeast and Fusarium in the south. Aspergillus is prevalent in both areas. Unlike bacterial keratitis, fungal keratitis tends to be a more indolent process. Also unlike bacterial keratitis, superficial corneal scrapings may be positive in up to 85% of cases. Fungal organisms tend to penetrate deep into the substance of the tissue rather than spreading along the surface or along the planes between corneal lamellae. Fungal organisms can readily penetrate through an intact Descemet’s membrane into the anterior chamber, causing a hypopyon early in the course of the disease, even before episcleral tissue becomes clinically inflamed. Characteristically, topical steroids are used before the organism becomes established in the corneal tissue121 (Fig. 22).
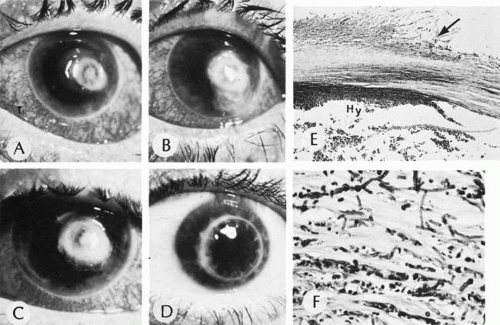 Fig. 22. Mycotic ulcer. A, B, and C. Progression of a fungal corneal ulcer. D. Same eye after a penetrating graft. E. Note the ragged appearance of the stromal lamellae (arrow) and the heavy neutrophilic infiltration at the edge of the ulcer. The organisms are most easily found and most viable at the periphery of the ulcer. Hy, hypopyon. F. High magnification of the edge of the ulcer. The stromal lamellae are infiltrated with neutrophils and invaded by branching, septate hyphae of the mold. (A, B, C, and D, clinical [AFIP negs. 73-10965, 73-10966, 73-10963, and 73-10967]; E, H&E, ×35 [AFIP] Acc. 831164]; F, PAS, ×400 [AFIP Acc. 831164]. A, B, C, and D modified from Zimmerman LE: Keratomycosis. Surv Ophthalmol 8:1, 1963; E and F, Fine BS: In King JH, McTigue JW [eds]: In The Cornea. Washington, DC, Butterworth & Co, 1965)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|