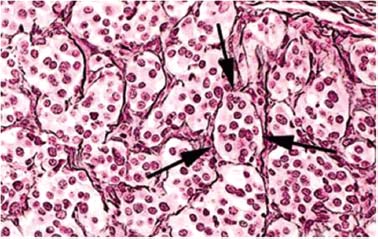
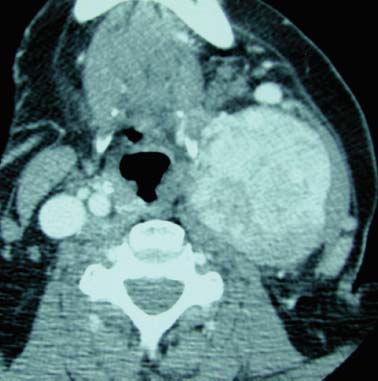
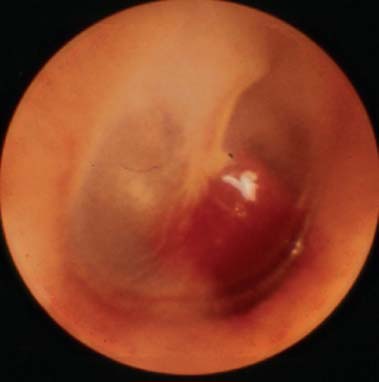
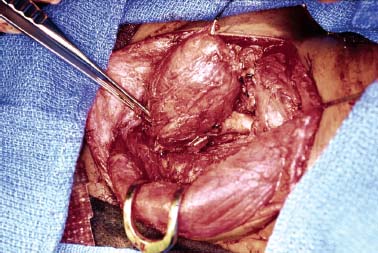
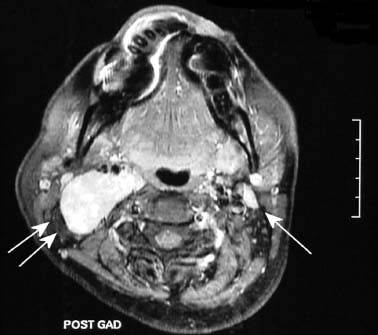
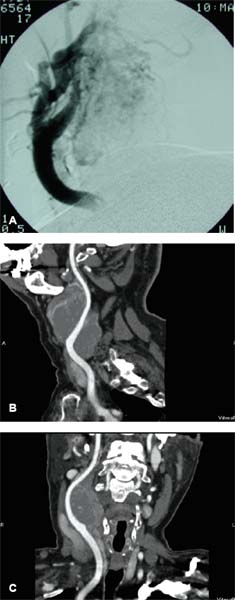
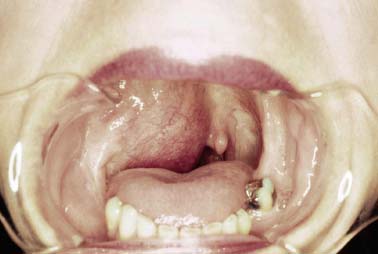
9 Core Messages • Paragangliomas are slow-growing neuroendocrine tumors that can exhibit multicentricity, rare biochemical activity, uncommon malignancy, and may be genetically transmitted. • About 10 to 50% are familial paragangliomas. • Only 3% of paragangliomas are biochemically active so testing for catecholamines should be reserved for patients with hypertension, tachycardia, or flushing. • Multicentricity may be seen in up to 20% of sporadic cases compared with 50% of familial paragangliomas. • On imaging, carotid paragangliomas can be distinguished from vagal paragangliomas by posterior and anterior displacement of the internal carotid artery. • Excision of paragangliomas exceeding 3 cm probably benefits from embolization. • Somatostatin-receptor scintigraphy allows for confirmation of paraganglioma location, identifying multiple tumors, and screening for familial paraganglioma and recurrence. • A team approach to the management of these tumors is strongly recommended. • Factors included in determining the best management of a paraganglioma include age, comorbidity, size of the mass, and existing cranial neuropathies. • Lymph nodes encountered during paraganglioma resection should be sent to pathology to rule out evidence of metastasis. • When laryngeal paraganglioma is suspected, a contrast computed tomography scan is recommended and transmucosal biopsy should be avoided to prevent excessive bleeding and mucosal scarring. Paraganglionic tissue was first described by Von Haller in 1743, but it was not until 1862 that the first carotid paraganglioma was described.1 Scudder excised the first carotid paraganglioma in 1903. These tumors are classically described as slow growing. They have the potential for multicentricity, hereditary transmission, biochemical activity, and malignancy. Paraganglionic cells are neural crest-derived cells that can be found from the base of the skull to the pelvis. These cells are part of the diffuse neuroendocrine system and are associated with nerves that have sympathetic systems. In the head and neck, paragangliomas are found along major vessels and the vagus nerve. Specific paraganglion cell sites are the carotid body, jugular bulb, Jacobsen’s tympanic plexus, vagus nerve, and supraglottic and infraglottic sites. The anatomic location of these slow-growing tumors gives rise to their more modern names (carotid paraganglioma, vagal paraganglioma, and jugular paraganglioma). The aforementioned were previously called carotid body tumors, glomus vagale, and glomus jugulare. Glomus tumors are of blood vessel origin, not neural crest derivation, and therefore the term glomus is a misnomer and is misleading. Paragangliomas have features that make them fascinating and determine their management. Anywhere from 10 to 50% are familially inherited, depending on where the study was performed.1 They are generally benign and slow growing and uncommonly are biochemically active. Multicentricity is more common in the familial variety. Management may be observation, surgical, or radiotherapeutical. The prevalence of paragangliomas is higher in communities living at high altitudes.2,3 Persons living at altitudes higher than 2000 m have been found to have an increased incidence of paraganglioma.3 Jech et al found no succinate dehydrogenase subunit B (SDHB) or SDH subunit D (SDHD) mutation in a small series of high-altitude paragangliomas.4 A similar increase in prevalence of paragangliomas in the Alps has not been observed.5 Some centers see more familial paraganglioma patients, increasing the number of patients seen and the percentage of familial patients evaluated. The most common paraganglioma is the carotid paraganglioma followed by the jugulotympanic paraganglioma. Tumor growth has been estimated to be 1.0 mm per year for actively growing tumors.6 In the same study including 48 tumors at three sites (carotid paraganglioma, vagal paraganglioma, and jugulotympanic paragangliomas), the doubling times for these sites were 7.13, 8.89, and 13.8 years, respectively. Forty percent of the tumors had no growth while being observed, and 81% of the tumors had a doubling time of greater than 3 years. Symptoms of paragangliomas are directly dependent on the site of the tumor, size of the tumor, and biochemical activity, if any. Therefore, tumor progression is uncommon, and observation in asymptomatic patients may be a reasonable treatment plan. Grossly, paragangliomas are rubbery vascular masses, usually densely adherent to surrounding structures, especially larger tumors. Carotid paragangliomas splay the internal and external carotid arteries, but they arise from the carotid body, which is located on the deep side of the bifurcation. The vagal paraganglions are found on the deep surface of the internal carotid artery, intermittently involving the vagus nerve. The tumors vary from gray to dark red. Microscopically, the hallmark of paraganglioma is Zellballen (Fig. 9.1). Chief cells and sustentacular cells are arranged in balls of cells. The central cells of the cell ball are polygonal. The surrounding cells are eosinophilic with occasional basophilic sustentacular cells. The chief cells are the active cells of the paraganglioma containing intracytoplasmic dense core granules. Immunostaining with chromogranin, synaptophysin, neuron-specific enolase, and neurofilament stain is usually positive, whereas immunostaining with mucicarmine, periodic acid-Schiff, and argentaffin stains is negative. Biological activity is present in less than 3% of the patients with head and neck paragangliomas.7 The paraganglioma chief cells lack N-methyl transferase and cannot convert norepinephrine to epinephrine, making these tumors chemically inactive. A fivefold increase in catecholamine levels is required for patients to experience palpitations, headache, sweating, and flushing. Patients with biochemically active tumors will have hypertension, and they should have a 24-hour urine test for vanillyl mandelic acid and metanephrines. In addition, further studies should be carried out to rule out associated pheochromocytomas. Multicentricity has been estimated to be present in 10 to 20% of sporadic cases.8 In a review of 1000 carotid paragangliomas, 31% of the cases were found to be bilateral if the patients had familial paragangliomas as opposed to 4% in sporadic cases.9 Multicentricity is estimated to be present in 10 to 50% of familial cases.9,10 In a study by Netterville et al on vagal paragangliomas, 80% of the cases were multicentric.11 Multiple paragangliomas should prompt a search for a familial origin in these tumors. It is possible that the variation in these numbers depends on the prevalence of familial paragangliomas in the study performed because multicentricity is a feature of familial tumors. Figure 9.1 A micrograph of Zellballen, the organoid arrangement of chief cells and sustentacular cells that make up paraganglionic tissue. Head and neck paragangliomas may present as sporadic or familial varieties (Table 9.1). Familial forms should be sought for several reasons. The hallmarks of a familial paraganglioma are known family member with a paraganglioma, multiple paragangliomas, younger age at onset/detection, male sex, and vagal paragangliomas.12 If a patient has the familial form, he or she is at risk for multiple tumors, which changes the treatment strategy. It promotes genetic screening for family members, allowing for earlier detection and treatment. One genetic variant (SDHB vide infra) is associated with a higher rate of pheochromocytomas and an increased incidence of malignant paragangliomas. Some paragangliomas have been associated with other tumor syndromes. MEN 2, von Hippel-Lindau disease, and neurofibromatosis type 1 have been linked to an increased risk of pheochromocytoma development.13 The gene(s) for the familial variety of paraganglioma has been localized to chromosome 11q23.14 Alterations in the SDH subunits (A, B, C, and D) result in tumor generation. The most common alteration is in SDHD for PGL1.15 PGL1 is inherited in a Mendelian fashion with genomic imprinting. In this instance, the paternally activated gene, if passed on, will give rise to a phenotypically positive offspring. During oogenesis, the gene is inactivated, resulting in no phenotypically positive children. However, a male carrier of an inactivated PGL1 gene will pass this gene on in a classic Mendelian fashion. The second most common PGL syndrome is the SDHB mutation. These cases have no parent-of-origin heritage and are more prone to malignancy.16 Germline mutations of SDHB, SDHC, and SDHD have been found in 30% of the sporadic cases.13 Despite the lack of familial characteristics, this suggests that all patients with paragangliomas should be screened for paraganglioma syndromes. Typically, the patient with a head and neck paraganglioma presents with a slow-growing, painless neck mass. Because paragangliomas are relatively rare, they are often not suspected in the initial evaluation of the patient with a neck mass. Unless very large or biochemically active, these tumors do not produce symptoms; therefore, a high index of suspicion is required to diagnose these lesions. The average duration from first detection to correct diagnosis is commonly quoted at 4 years. Probably the most common presentation in patients with sporadic paragangliomas is either a positive computed tomography (CT) scan obtained for the evaluation of the neck mass or an incidental finding in patients getting duplex Doppler scans to evaluate carotid stenosis (Fig. 9.2). Notation about the sex of the patient and age at onset may be helpful in determining whether the patient has a familial paraganglioma. A family history of paraganglioma should be sought. An earlier treatment for a paraganglioma or multiple tumors strongly suggests a familial tumor. Keep in mind that a history of pheochromocytoma is also strongly suggestive of one of the paraganglioma syndromes. The patient’s place of origin (endemic areas or high-altitude dwelling) must be considered.17 Symptoms of hearing loss, tinnitus, cranial neuropathy, Horner syndrome, dysphagia, and dysphonia need to be elicited. They may be subtle with moderate-sized tumors. Headaches, palpitations, flushing, and sweating may be signs of biochemical activity. A history of elevated heart rate and hypertension must be obtained in every patient. Figure 9.2 Typical appearance of a carotid paraganglioma. Physical examination of all regions of the head and neck should be performed. Tympanic paragangliomas will present with a red mass behind the tympanic membrane (Fig. 9.3). Brown sign is a blanching of the tympanic paraganglioma when pneumotoscopic pressure is placed on the tympanic membrane. With carotid paraganglioma, the mass can be moved in the anterior/posterior plane but not in the craniocaudad plane. These authors find bruits to be uncommon, but the carotid pulse is transmitted through the mass. When larger, carotid paragangliomas have findings similar to those of parapharyngeal space tumors: unilateral serous otitis, snoring, and bulging of the soft palate (Fig. 9.4). Figure 9.3 Tympanic paraganglioma. The nonotologic head and neck paragangliomas can all give rise to cranial nerve damage. Cranial nerve involvement is a late sign (cranial nerves IX through XII). Dysphagia and/or hoarseness can occur with larger carotid tumors. The larger the paraganglioma, the more likely pretreatment neuropathy will be present. Dysphagia can be subtle and due to neural involvement or mass effect. Vagal tumors arise higher in the parapharyngeal space and frequently involve cranial nerves at the time of presentation (Fig. 9.5). In vagal paragangliomas, the vagus nerve is involved early and the site of involvement can be determined by physical examination. Masses at the nodose ganglion will paralyze the vocal fold and spare the soft palate. After getting the history and physical examination, imaging should be obtained. The goals of imaging are to determine the size, location, relationship to the carotid artery, discover multiple paragangliomas, and reveal local invasion/destruction, especially at the skull base. Figure 9.5 Right vagal paraganglioma. Note the position of the underlying carotid artery. Imaging plays a critical role in the diagnosis, management, and surveillance of these tumors. The patient suspected of having a paraganglioma may be evaluated by CT, magnetic resonance imaging (MRI), and/or the angiographic equivalent of these modalities. CT scanning can provide critical information about size, location, multicentricity, and local tissue involvement (Fig. 9.6). It is probably not as sensitive as MRI at the skull base and with regard to local tissue involvement. CT scanning with contrast can reveal an enhancing mass splaying the internal and external carotid arteries in carotid paragangliomas. The vagal paragangliomas will displace the internal carotid artery anteriorly, while the carotid paragangliomas will displace it posteriorly (Fig. 9.7A, B). CT scanning of the temporal bone demonstrates the bony anatomy of the middle ear and jugular foramen.18 Tympanic paragangliomas appear as soft-tissue masses on the cochlear promontory. Ossicular destruction is evident as these tumors grow. Jugular paragangliomas destroy by erosion, expanding the jugular fossa. Larger tumors erode into the eustachian tube, carotid canal, and neuroforamina (Fig. 9.8). Very large lesions will destroy the jugular spine and the hypoglossal canal, and finally will invade the cranial contents. Flow voids within paragangliomas create the classic “salt and pepper” appearance on MRI (Fig. 9.9). Dural involvement and skull base involvement are best detected with gadolinium-enhanced MRI. Figure 9.6 Magnetic resonance imaging of a right carotid paraganglioma. If a lesion is detected, further confirmation can be obtained with 111Indium pentetreotide (octreotide) imaging (Fig. 9.10). Octreotide is a radiolabeled somatostatin analog that targets receptor-rich cells. It differs from positron emission tomography (PET) imaging in that it is a measure of receptor density rather than cell activity. Published sensitivity of this scan is in excess of 90%.19 It is also beneficial in determining multifocal tumors, metastases from malignant paragangliomas, and has been used as a screening method for familial paragangliomas.20,21 The latter has been supplanted by genetic testing. 18-Fluorine L-3, 4-dihydroxyphenylalanine (18F-DOPA) PET has been shown to be an even more sensitive modality than octreotide. It is more extensive as well. Imaging of neuroendocrine tumors with 18F-DOPA PET has an accuracy of 90%.22 Hoegerle et al studied paragangliomas with 18F-DOPA PET.23 They demonstrated the superiority of 18F-DOPA PET scanning in detecting small paragangliomas that could be missed on MRI. Both these modalities have replaced metaiodobenzylguanidine (MIBG) imaging for paragangliomas. Figure 9.7 (A) This carotid paraganglioma has displaced the internal carotid artery posteriorly. (B, C) These vagal paragangliomas have displaced the internal carotid artery anteriorly.
Paragangliomas of the Head and Neck
Natural Course of Disease
Hereditary Paragangliomas

History and Physical Examination
Imaging
Paragangliomas of the Head and Neck
Only gold members can continue reading. Log In or Register to continue

Full access? Get Clinical Tree


