Purpose
To evaluate the ocular hypotensive efficacy of 0.05%, 0.1% and 0.25% AR-12286 Ophthalmic Solutions in patients diagnosed with ocular hypertension or glaucoma.
Design
Parallel comparison, vehicle-controlled, double-masked, 3-week randomized clinical trial.
Methods
Subjects (n = 89) with elevated intraocular pressure (IOP) were assigned randomly to receive either 1 of 3 concentrations of AR-12286 or its vehicle. Dosing was once-daily in the morning for 7 days, then once-daily in the evening for 7 days, then twice daily for 7 days. Primary and secondary efficacy end points were mean IOP at each diurnal time point (8 am, 10 am, 12 pm, and 4 pm) and mean change in IOP from baseline, respectively.
Results
All 3 concentrations of AR-12286 produced statistically and clinically significant reductions in mean IOP that were dose dependent, with peak effects occurring 2 to 4 hours after dosing. Mean IOP at peak effect ranged from 17.6 to 18.7 mm Hg (−6.8 to −4.4 mm Hg) for the 3 concentrations. The largest IOP reductions were produced by 0.25% AR-12286 after twice daily dosing (up to −6.8 mm Hg; 28%). The 0.25% concentration dosed once-daily in the evening produced highly significant IOP reductions throughout the following day (−5.4 to −4.2 mm Hg). The only adverse event of note was trace (+0.5) to moderate (+2) conjunctival hyperemia that was transient, typically lasting 4 hours or less. After once-daily evening dosing, hyperemia was seen in less than 10% of patients.
Conclusions
AR-12286 was well tolerated and provided clinically and statistically significant ocular hypotensive efficacy in patients with ocular hypertension and glaucoma.
Glaucoma is a progressive optic neuropathy that causes characteristic loss of visual fields and eventually can lead to blindness. A major risk factor for glaucomatous visual field loss is elevated intraocular pressure (IOP). Studies such as the Early Manifest Glaucoma Trial, the Ocular Hypertension Treatment Study, and the Collaborative Normal Tension Glaucoma Study support the role of lowering IOP in reducing the risk of glaucomatous progression in patients with ocular hypertension and glaucoma. For many patients, however, current glaucoma medications are not sufficiently effective as monotherapy to achieve low target IOPs, and approximately half of patients with elevated IOP are treated by coadministration of 2 or more glaucoma medications. Thus, there remains a need for novel pharmacotherapies that can produce greater IOP-lowering efficacy with improved dosing convenience for patients.
Most currently used glaucoma medications reduce IOP either by reducing the flow of aqueous humor into the eye or by improving the drainage of this fluid through the uveoscleral pathway. Rho kinase inhibitors have been shown to reduce IOP in rabbits and monkeys by increasing aqueous humor drainage through the primary outflow pathway in the eye, the trabecular meshwork. Several lines of experimental evidence indicate that modulating the activity of Rho kinase within the primary aqueous humor outflow pathway may offer substantial advantages for the treatment of patients with glaucoma. In addition to improving the outflow function of the trabecular meshwork, Rho kinase inhibitors have been shown to increase ocular blood flow and to enhance retinal ganglion cell survival after ischemic injury.
AR-12286 is a novel, potent Rho kinase inhibitor with single-digit nanomolar inhibitory activity against Rho kinase in enzymatic inhibition assays (deLong MA, et al. IOVS 2009;50:ARVO E-abstract 4058). Mechanism-of-action studies in monkeys demonstrate that AR-12286 lowers IOP primarily by increasing aqueous humor outflow through the trabecular meshwork (Wang RF, et al. IOVS 2009;50:ARVO E-abstract 1465). The objectives of this study were to evaluate the ocular hypotensive efficacy along with the ocular and systemic safety of AR-12286 ophthalmic solutions.
Methods
In this parallel comparison, vehicle-controlled, double-masked study, subjects were randomized to receive either 1 of 3 concentrations of formulated AR-12286 ophthalmic solution or its vehicle (preserved with benzalkonium chloride 0.015%), packaged in identical containers. Dosing was once-daily in the morning for 7 days, then once-daily in the evening for 7 days, then twice daily for 7 days. This dosing regimen was used to evaluate whether twice-daily dosing was more effective than once-daily dosing. Morning versus evening dosing for the once-daily regimen was evaluated to evaluate if this new class of agents was similar to prostaglandins, where evening dosing has been reported to be more effective. Dosing on days 1, 2, 8, and 22 was performed in the clinic, and on other days it was self-administered (7 am to 9 am and 8 pm to 10 pm as appropriate).
To be included in the study, the subjects were required to be 18 years of age or older with a diagnosis of open-angle glaucoma or ocular hypertension. The entry requirements for unmedicated IOP were IOP of 24 mm Hg or more in 1 or both eyes at 8 am and of 21 mm Hg or more at 10 am, 12 pm, and 4 pm at the screening visit. Other entry requirements included corrected visual acuity in each eye of +1.0 logarithm of the minimal angle of resolution or units better by Early Treatment Diabetic Retinopathy Study charts in each eye (equivalent to 20/200) and the ability and willingness to give signed informed consent and to follow study instructions. Excluded from the study were individuals with IOP of more than 36 mm Hg; recent ocular trauma, surgery, infection, or inflammation; or concomitant ocular medications. Also excluded were patients with central corneal thickness of more than 600 μm or those who had undergone previous glaucoma incisional or laser surgery or refractive procedures. Because of the status of the preclinical safety program, women of childbearing potential who were pregnant, nursing, planning a pregnancy, or not using a medically acceptable form of birth control were excluded.
The screening examination included seated measurement of heart rate and blood pressure, complete blood count and chemistry panel assessment, visual acuity (Early Treatment Diabetic Retinopathy Study charts), IOP, biomicroscopy (using a 0-to-3 scale for none to severe), dilated ophthalmoscopy, and automated threshold visual fields (within 3 months). Qualified individuals using topical ocular hypotensive underwent a washout as follows: prostaglandins and β-adrenoceptor antagonists (4 weeks), adrenergic agonists (2 weeks), and muscarinic agonists and carbonic anhydrase inhibitors (5 days). Subsequent examinations included visual acuity, IOP, and biomicroscopy, with dilated ophthalmoscopy at the final visit (day 23).
The primary efficacy end point was the mean IOP across subjects within the treatment group on each day at each posttreatment time point. Secondary efficacy end points included: mean change from diurnally adjusted baseline IOP at each time point, mean percent change from diurnally adjusted baseline IOP at each time point, mean diurnal IOP on each day with 4 diurnal IOP measurements, and mean change from the baseline mean diurnal IOP on each day with 4 diurnal IOP measurements. A priori, the primary population selected for efficacy was all patients who received at least 1 dose of the study medication and had 1 follow-up visit (modified intent-to-treat population). If 2 eyes were qualified, the worse eye (ie, the eye with the higher IOP at the screening visit) was chosen as the study eye. If the IOP measurements were the same, the right eye was the study eye. Only 1 eye was treated and analyzed. No corrections for multiplicity were made because of the exploratory nature of this early study. All safety analyses were carried out using the safety population (all randomized subjects who received at least 1 dose of study medication). Differences across treatment groups in incidence of adverse events were tested using a Fisher exact test. Paired t tests were used to test within-treatment group changes for vital sign parameters (PC-SAS software version 9; SAS Institute, Cary North Carolina, USA). A priori, a sample size of 20 per group provided 80% power to detect a difference of 3.5 mm Hg or more between each dose and its vehicle (α = 0.05, 1-tailed for this vehicle-controlled study).
Results
Disposition and Demographics
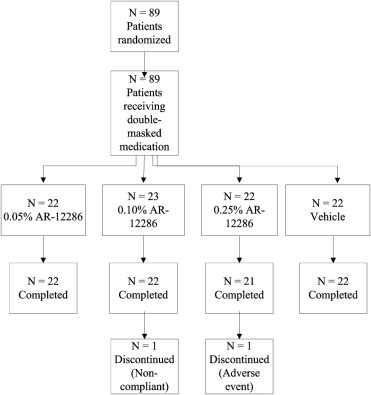
A total of 89 individuals were enrolled as study patients, of which 87 patients completed the study ( Figure 1 ). One noncompleting patient was in the 0.10% AR-12286 treatment group (noncompliant) and the other was in the 0.25% AR-12286 group (adverse event, hyperemia). The safety population included all 89 patients, as did the modified intent-to-treat population.
The prestudy characteristics of the population are shown in Table 1 . There were no clinically or statistically significant differences among the treatment groups. Approximately half of the patients required a washout of ocular hypotensive medications (51.7%; 46/89). Of the 46 patients using ocular hypotensive medications before the study, 32 patients (70%) were using a prostaglandin analog.
| Characteristics | AR-12286 0.05% (n = 22) | AR-12286 0.10% (n = 23) | AR-12286 0.25% (n = 22) | Solution Vehicle (n = 22) | All Treatments (n = 89) | P Value a |
|---|---|---|---|---|---|---|
| Sex, n (%) | ||||||
| Male | 11 (50.0) | 12 (52.2) | 8 (36.4) | 8 (36.4) | 39 (43.8) | .589 |
| Female | 11 (50.0) | 11 (47.8) | 14 (63.6) | 14 (63.6) | 50 (56.2) | |
| Age (y) | ||||||
| Mean ± SD | 67.3 ± 10.42 | 65.1 ± 9.86 | 64.8 ± 9.93 | 60.5 ± 10.60 | 64.4 ± 10.32 | .175 |
| Range | 49 to 85 | 46 to 81 | 43 to 80 | 36 to 77 | 36 to 85 | |
| Race, n (%) | ||||||
| Asian | 0 (0.0) | 1 (4.3) | 0 (0.0) | 0 (0.0) | 1 (1.1) | .167 |
| Black | 2 (9.1) | 2 (8.7) | 7 (31.8) | 5 (22.7) | 16 (18.0) | |
| White | 20 (90.9) | 20 (87.0) | 15 (68.2) | 17 (77.3) | 72 (80.9) | |
| Ethnicity, n (%) | ||||||
| Hispanic | 4 (18.2) | 3 (13.0) | 4 (18.2) | 3 (13.6) | 14 (15.7) | .940 |
| Not Hispanic | 18 (81.8) | 20 (87.0) | 18 (81.8) | 19 (86.4) | 75 (84.3) | |
| Iris color, n (%) | ||||||
| Brown | 11 (50.0) | 12 (52.2) | 16 (72.7) | 11 (50.0) | 50 (56.2) | .459 |
| Hazel | 3 (13.6) | 4 (17.4) | 4 (18.2) | 2 (9.1) | 13 (14.6) | |
| Blue | 6 (27.3) | 5 (21.7) | 2 (9.1) | 7 (31.8) | 20 (22.5) | |
| Green | 2 (9.1) | 2 (8.7) | 0 (0.0) | 0 (0.0) | 4 (4.5) | |
| Black | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (4.5) | 1 (1.1) | |
| Other | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (4.5) | 1 (1.1) | |
| Mean central corneal thickness (μm) | ||||||
| Mean ± SD | 564.80 ± 22.936 | 558.07 ± 23.325 | 546.23 ± 25.434 | 553.39 ± 36.055 | 555.65 ± 27.786 | .155 |
| Glaucoma history | ||||||
| Open-angle glaucoma | 12 (54.5) | 13 (56.5) | 17 (77.3) | 10 (45.5) | 52 (58.4) | .181 |
| Ocular hypertension | 10 (45.5) | 10 (43.5) | 5 (22.7) | 12 (54.5) | 37 (41.6) | .181 |
| IOP (mm Hg) | ||||||
| Screening | ||||||
| Mean ± SD | 20.1 ± 4.38 | 21.9 ± 4.55 | 21.9 ± 3.94 | 21.4 ± 4.92 | 21.3 ± 4.45 | .519 |
| Baseline | ||||||
| Mean ± SD | 26.0 ± 2.17 | 27.3 ± 3.18 | 26.9 ± 2.03 | 26.3 ± 2.47 | 26.6 ± 2.52 | .330 |
| Washout of prestudy medications required? | ||||||
| No | 12 (54.5) | 11 (47.8) | 10 (45.5) | 10 (45.5) | 43 (48.3) | — |
| Yes | 10 (45.5) | 12 (52.2) | 12 (54.5) | 12 (54.5) | 46 (51.7) |
a Tests of differences across the treatment groups and 2-sided. Categorical responses are tested using the Fisher exact test. Tests of differences in means are performed using a 1-way analysis of variance model including treatment as the only between-group effect.
Efficacy
Over the course of the unmedicated, baseline diurnal day (day 1), mean IOP in the study eyes ranged from 26.0 to 27.3 mm Hg between groups at 8 am (P = .330) and decreased to 23.7 to 24.2 mm Hg by 4 pm. All 3 concentrations of AR-12286 produced statistically and clinically significant (P < .001) reductions in mean IOP relative to baseline, with peak effects occurring 2 to 4 hours after acute or chronic dosing ( Figures 2 and 3 ). The overall hypotensive efficacy was dose dependent, with the 0.25% concentration producing the largest reductions in mean IOP (6.8 mm Hg) and the longest duration of effect, up to 24 hours after a once-daily dose. Twice-daily dosing of 0.25% AR-12286 provided a greater ocular hypotensive efficacy throughout the day than once-daily dosing in the morning or the evening ( Figures 3 and 4 ). Once-daily dosing of the 0.25% concentration in the evening produced highly significant reductions in mean IOP throughout the following day, ranging from 19.6 to 21.5 mm Hg (−5.4 to −4.2 mm Hg). With the exception of 8 am on day 8, the 0.25% AR-12286 treatment group was statistically significantly different from vehicle (P < .05) at all posttreatment visits.
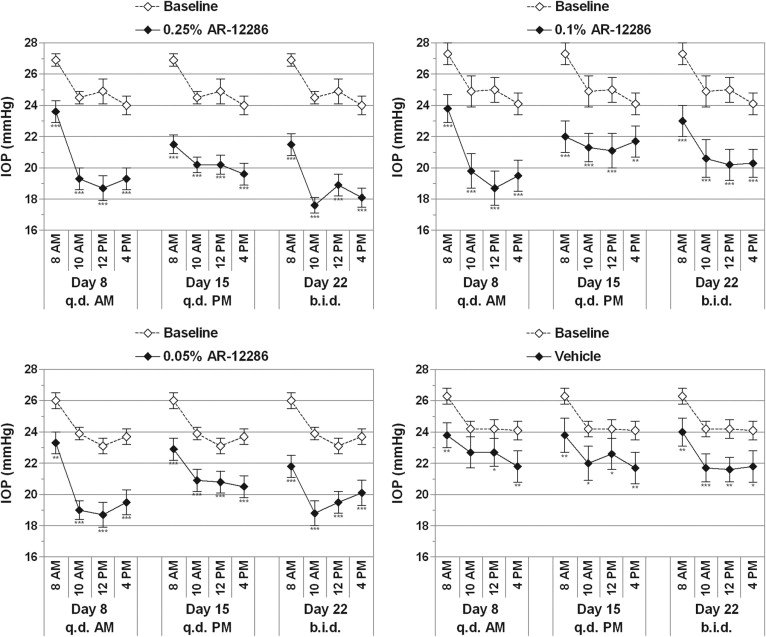
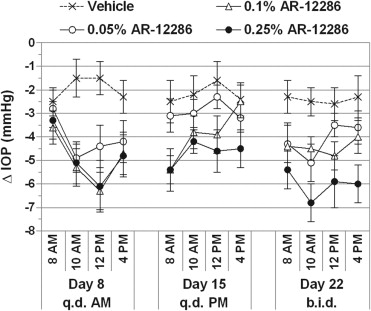
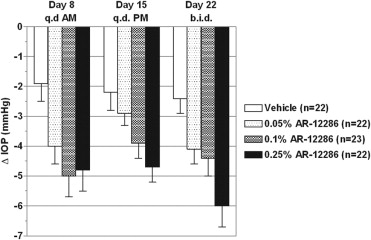
Changes in IOP from baseline were dose dependent and statistically significant for all AR-12286 concentrations and vehicle ( P ≤ .011). However, the 0.25% AR-12286 concentration was the only treatment to achieve the highest level of statistical significance ( P < .001) at all posttreatment time points. There were no differences of note between the modified intent-to-treat and per protocol (PP) analyses of efficacy. A planned stratification of efficacy by iris color, age, gender, and race found no obvious efficacy differences between demographic subgroups.
Safety
Ninety-one adverse events were reported in a total of 49 patients: 22 in the AR-12286 0.05% group (13/22 patients; 59.1%), 27 in the AR-12286 0.10% group (13/23 patients; 56.5%), 32 in the AR-12286 0.25% group (16/22 patients; 72.7%), and 10 in the vehicle group (7/22 patients; 31.8%). The severity of events was reported to be mild (34/49; 69.4%) or moderate (15/49; 30.6%), with none reported to be severe. Most of the events (79/91; 86.8%) were judged by the investigator to be related to the study medication. The most frequently reported events were in the eye disorders organ system, with 31.8% (7/22), 47.8% (11/23), 63.6% (14/22), and 9.1% (2/22) for the 0.05%, 0.10%, and 0.25% AR-12286 and vehicle groups, respectively. The most frequently reported events were ocular hyperemia and conjunctival hyperemia. Together, the incidence of hyperemia was 27.3% (6/22), 39.1% (9/23), 59.1% (13/22), and 9.1% (2/22) for the 0.05%, 0.10%, and 0.25% AR-12286 and vehicle groups, respectively, suggesting a dose relationship. Other events, ocular or systemic, were relatively infrequent and similar in active and vehicle treatment groups ( Table 2 ). There were no serious adverse events and no safety issues regarding the untreated fellow eye.



