Neurofibromatosis type 2 (NF2) is a rare syndrome characterized by bilateral vestibular schwannomas, multiple meningiomas, cranial nerve tumors, spinal tumors, and eye abnormalities. NF2 presents unique challenges to the otologist because hearing loss may be the presenting complaint leading to the diagnosis of the disorder. Care of patients with NF2 requires knowledge of all tumors and symptoms involved with the disorder. It is recommended that patients receive care in a center with expertise in NF2. The role of the neuro-otologist in this care is determined by the specialty center.
Key points
- •
Care of patients with neurofibromatosis type 2 (NF2) requires knowledge of all tumors and symptoms involved with the disorder.
- •
It is recommended that patients receive care in a center with expertise in NF2.
- •
The role of the neuro-otologist in this care is determined by the specialty center.
Introduction
Neurofibromatosis (NF) is a rare syndrome characterized by bilateral vestibular schwannomas, multiple meningiomas, cranial nerve (CN) tumors, spinal tumors, and eye abnormalities. Neurofibromatosis type 2 (NF2) presents unique challenges to the otologist because hearing loss may be the presenting complaint leading to the diagnosis of the disorder. NF2 is invasive, requiring a multispecialist team approach for the evaluation and treatment of the disorder. The primary impairment is hearing loss resulting from bilateral vestibular schwannomas. NF2 must be differentiated from neurofibromatosis type 1 (NF1); although the names are linked, the disease entities are distinctly different. This article reviews the clinical characteristics of NF2 and current recommendations for evaluation and treatment.
Introduction
Neurofibromatosis (NF) is a rare syndrome characterized by bilateral vestibular schwannomas, multiple meningiomas, cranial nerve (CN) tumors, spinal tumors, and eye abnormalities. Neurofibromatosis type 2 (NF2) presents unique challenges to the otologist because hearing loss may be the presenting complaint leading to the diagnosis of the disorder. NF2 is invasive, requiring a multispecialist team approach for the evaluation and treatment of the disorder. The primary impairment is hearing loss resulting from bilateral vestibular schwannomas. NF2 must be differentiated from neurofibromatosis type 1 (NF1); although the names are linked, the disease entities are distinctly different. This article reviews the clinical characteristics of NF2 and current recommendations for evaluation and treatment.
Neurofibromatosis type 2 differentiated from neurofibromatosis type 1
NF1 has distinctly different characteristics from NF2. NF1 and NF2 have been distinguished as completely different genetic diseases based on the chromosome responsible for the disease. NF1 has been localized to chromosome 17, and NF2 has been localized to chromosome 22. NF1 is a multisystem disorder in which some features may be present at birth and others are age-related manifestations. The National Institutes of Health (NIH) Consensus Development Conference identified the following 7 features of the disease, of which 2 or more are required to establish the diagnosis of NF1:
- 1.
Six café au lait spots equal to or greater than 5 mm in longest diameter in prepubertal patients and 15 mm in longest diameter in postpubertal patients
- 2.
Two or more neurofibromas of any type or 1 plexiform neurofibroma
- 3.
Freckling in the axilla or inguinal regions
- 4.
Optic glioma (optic pathway glioma)
- 5.
Two or more Lisch nodules (iris, hamartoma)
- 6.
Distinct osseous lesions, such as sphenoid wing dysplasia or cortical thinning of the cortex of long bones with or without pseudoarthrosis
- 7.
First-degree relative (parent, sibling, or child) with NF1 according to the criteria listed earlier
Some patients also manifest learning disabilities or language disorders. A careful examination and a detailed history of the patient’s symptoms help distinguish NF1 and NF2.
Clinical characteristics of neurofibromatosis type 2
Definition
The NIH Consensus Development Conference also developed guidelines for the diagnosis of NF2. NF2 is distinguished by bilateral vestibular schwannomas with multiple meningiomas, CN tumors, optic gliomas, and spinal tumors. A definite diagnosis is made from the presence of bilateral vestibular schwannomas or developing a unilateral vestibular schwannoma by age 30 years and a first-degree blood relative with NF2, or the presence of a unilateral vestibular schwannoma and developing at least 2 of the following conditions known to be associated with NF2: meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacity/juvenile cortical cataract ( Box 1 ).
Confirmed (definite) NF2
Bilateral VS or family history of NF2 (first-degree family relative) plus
- 1.
Unilateral VS less than 30 years, or
- 2.
Any 2 of the following: meningioma, glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
- 1.
Presumptive or probable NF2: should evaluate for NF2
Unilateral VS less than 30 years plus at least 1 of the following:
Meningioma, glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
Multiple meningiomas (≥2) plus unilateral VS less than 30 years or one of the following:
Glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
Abbreviation: VS, vestibular schwannoma.
There may be significant heterogeneity in the presentation of the disease from one individual to the next. Some individuals may have a very mild form of the disease, such as small vestibular schwannomas manifesting in an older individual. At the other end of the spectrum, some children present with multiple intracranial tumors at a very young age. Despite the heterogeneity of the disease, the expression of NF2 tends to be similar within a family. There is a significant genetic component to the disease with much variability within the parameters of the observed phenotype. Studies have shown that a truncating mutation (nonsense and frame shift) may be linked to a more severe form of NF2. The more severe form of NF2 is termed the Wishart form. Individuals with this severe form present with early onset of the disease, with multiple intracranial schwannomas and meningiomas that result in blindness, deafness, paralysis, and possibly death by age 40 years. Despite the strong genotype-phenotype correlation seen in NF2, individual differences in tumor growth occur within patients, making it difficult to predict how an individual’s tumors will change over time even when the genotype is known. The milder form, the Gardner form, of NF2 is less debilitating. Schwannomas may remain stable for many years, few meningiomas develop, and patients may not develop symptoms until later in life and often have fewer associated disabilities. The genetic basis of the mild form has not been well characterized. Many of these may be mosaic forms of the disease.
Prevalence and Incidence
The average age of diagnosis of NF2 is 25 years; however, many patients present with symptoms before the diagnosis. There is an average delay of diagnosis of approximately 7 years. There is no difference in the proportion of men versus women who develop NF2, and no prevalence has been described based on ethnicity. Epidemiologic studies place the incidence of NF2 between 1 in 33,000 live births and 1 in 87,410 live births.
Imaging Studies
All patients suspected to have NF2 should have a high-quality MRI scan performed with thin cuts through the internal auditory canals (IAC). All patients diagnosed with a unilateral vestibular schwannoma should have a dedicated IAC series to ensure there is not another tumor on the opposite side. Patients diagnosed with NF2 should have a complete spine series to evaluate the spine and stage the disease. Small spinal tumors commonly may be found in the cauda equina area, and occasionally a large asymptomatic schwannoma or meningioma may be found in the spine that could require treatment. Early treatment of spine tumors can significantly reduce the morbidity associated with these lesions. It is unusual for patients older than 40 years to present with NF2, although with more sensitive MRI scanning techniques, more of these individuals are being diagnosed at an older age. Metastasis may rarely manifest with bilateral IAC lesions, and it is important that carcinoma be ruled out in any patient older than 40 years who presents with bilateral IAC lesions.
A patient identified with NF2 should have a complete cranial MRI scan with cervical thoracic and lumbar spinal imaging. This scan serves as a baseline. A 6-month follow-up MRI scan is recommended for the intracranial tumors. If the tumors show stability, a yearly MRI scan is performed on the intracranial structures. Large tumors in the spine may be monitored with a similar frequency. If no spinal tumors are present, spinal imaging should be performed if the patient becomes symptomatic. Monitoring spinal tumors every 3 years is recommended when these are present. Intracranial imaging is performed on a yearly basis unless studies over several years indicate stability of the tumors.
Molecular Genetics
The NF2 gene was mapped to chromosome 22q12–2 in 1993. The NF2 gene located at chromosome 22 codes for a tumor suppressive protein termed merlin or schwannomin. This protein negatively regulates Schwann cell production and the loss of this protein allows overproduction of Schwann cells. The mutation in the NF2 chain predisposes individuals to developing a schwannoma when the second hit occurs to the gene; control of Schwann cells is lost or mutated within the cell. Various types of mutations have been identified, including single-base substitutions, insertions, and deletions. The mild, or Gardner, type of NF2 may be associated with missense mutations, whereas associations between the other mutations and phenotypes are not as clear. The occurrence of NF2 is not restricted to families known to carry the mutation. Frequently, genetic mosaicism occurs, which may not be detected by common mutation analysis techniques. Unilateral vestibular schwannomas may show the same type of genetic markers as NF2. The mutations in unilateral vestibular schwannomas are confined to the affected tumor tissue. In patients with NF2, the mutation is present in all cell types.
Family History
NF2 is an autosomal dominant disease, and 50% of children of affected individuals are at risk for developing the disease. Of patients in whom NF2 is diagnosed, 50% present with a family history of NF2. Half of all NF2-affected patients have no family history of NF2 and are considered founder cases. NF2 presentation and phenotype tend to be similar within families. The likelihood of NF2 occurring in related individuals who do not show similar clinical symptoms to an affected family member is small. Consideration must still be given to screening these individuals for risks despite the lack of clinical symptoms. Individuals at risk for developing NF2 must be screened to provide an early diagnosis. Individuals at risk include children of NF2-affected patients and their siblings. Fifty percent of all children of patients with NF2 are found to have the disease. Siblings of a patient diagnosed with NF1 are at risk, especially if the parent also has NF2. The type of screening and timing of screening depend on each NF2 center’s preference. Early screening is advocated so that tumors may be diagnosed presymptomatically. Screening may occur via MRI or genetic blood testing.
Screening
MRI screening of potentially affected individuals uses a postcontrast T1-weighted sequence of the full head with thin cuts through the IAC. A dedicated IAC MRI scan identifies most patients with NF2 by showing any vestibular schwannomas. Screening of the spine and ophthalmologic examination should be considered if the cranial MRI scan is positive. An audiogram (pure tone thresholds) or current clinical standard auditory brainstem response (ABR) testing is likely to miss small vestibular schwannomas. MRI can diagnose presymptomatically.
MRI is recommended for at-risk children when this test can be performed without sedation; this usually can be done when the child is 7 to 9 years old. A recommended first step for children younger than 7 years is an audiogram. Any child with an NF2-associated symptom, such as hearing loss or facial weakness, should be screened without regard to the need for sedation or age; MRI should be performed as soon as possible after the symptoms become apparent.
Identification of the NF2 gene and chromosome 22 has made genetic testing possible. It is recommended that patients with NF2 see a genetic counselor to discuss the hereditary consequences of the disease. Blood testing for the mutation is able to identify the defect of the NF2 gene in approximately 70% to 75% of patients with a known diagnosis of NF2. If the defect is identified in the affected individual, potential family members may be screened. If the gene is not identified in the affected individuals, blood screening of family members cannot be performed. The use of blood screening for patients without a diagnosis of NF2 or with a suspected diagnosis of NF2 is not recommended. New mutations in patients with mild presentation are most likely to be missense mutations, and this is difficult to identify with genetic testing of patients with NF2. The best molecular testing is performed on fresh vestibular schwannoma tissue. Any patient with a planned vestibular schwannoma removal should be offered the opportunity to have their tumor tissue tested for the genetic mutation. This test is the most sensitive and allows identification of the genetic mutation in most cases. The results of this test can then be used to screen relatives for NF2. There are currently 2 clinical laboratories that perform this test (Massachusetts General and University of Alabama) and there are many research laboratories associated with NF centers that may perform the test on recently resected tissue. The tissue must be keep frozen and testing should be coordinated with the laboratories before surgical resection.
Schwannoma tumor types
Bilateral vestibular schwannomas (acoustic neuromas) are benign neoplasms of the acoustic or eighth CN ( Fig. 1 ). The tumors are thought to arise at the glioma–Schwann cell junction within the internal auditory meatus. The tumors most commonly arise from the superior vestibular nerve, although, with NF2, tumors may be found on the cochlear and facial nerves within the internal auditory meatus. The consequences of a vestibular schwannoma are numerous, including hearing loss progressing to deafness; dizziness and balance problems; tinnitus; facial nerve paralysis; brainstem compression; and, if left untreated, death.
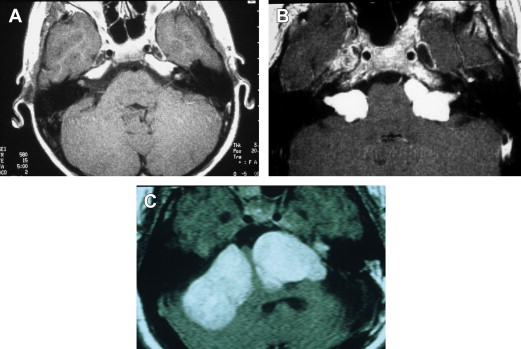
Despite the strong genetic effect in NF2, there is enormous variability in the number of tumor types, the rate of progression, and the disabilities experienced. This enormous variability is also found in patient presentation ( Table 1 ). Some patients may be asymptomatic. Patients who have no symptoms when diagnosed have generally been identified from genetic analysis conducted because a blood relative has NF2 or presymptomatic screening.
| Symptoms | Patients (%) |
|---|---|
| Neurologic | 17.5 |
| Skin tumor | 11.7 |
| Vision loss | 10.7 |
| Asymptomatic | 10.7 |
| Tinnitus | 7.8 |
| Weakness | 2.9 |
| Vertigo | 1 |
| Other/unspecified | 4.9 |
Although the NIH criteria for NF2 require the presence of bilateral vestibular schwannomas for diagnosis, patients may first develop unilateral schwannomas as a young child with no other tumors, or adult patients may present with multiple meningiomas (cranial and spinal) and no vestibular schwannomas. Although the NIH criteria for NF2 imply that all patients with NF2 develop bilateral vestibular schwannomas, some researchers are not convinced of this. Evans and colleagues based their conclusion on the observation of a possible variant form of NF2 manifesting with skin and spinal tumors in the absence of vestibular schwannomas. Nonetheless, the phenotype generally is reflective of the underlying disorder.
The natural history study of vestibular schwannomas in NF2 conducted at the House Ear Institute showed that 10 of 80 (12.5%) enrolled subjects had no symptoms at diagnosis, and 23 (28.8%) had cranial meningiomas and spinal meningiomas in addition to bilateral vestibular schwannomas. Nearly half (47.5%) had 1 vestibular schwannoma removed before enrollment. In general, the tumor resected before enrollment was removed 1.5 years after discovery, and was an average of 2.1 cm at removal. Few patients in the natural history study had spinal tumors or meningiomas removed before enrollment. The preliminary data indicate that, for this sample of subjects with NF2, the most salient medical issue is the growth of the vestibular schwannomas.
Intracranial Schwannomas
Vestibular schwannomas are the most common intracranial schwannomas associated with NF2. The most frequently identified nonvestibular schwannomas are schwannomas of CN III and V. Bilateral CN III or V schwannomas are the most common additional schwannomas seen. It is important to identify these lesions on MRI. Lower CN schwannomas may also be identified, but are much less frequently seen. A vestibular schwannoma rarely turns malignant, and sometimes unilateral vestibular schwannomas regress in size altogether. Growth of the tumors does not seem to be related either to loss of heterozygosity (genetic level of analysis) or to auditory functioning (phenotype level of analysis). For this reason, it is recommended that a patient have at least yearly MRI scans to track changes in size. All newly diagnosed patients should have a full head and spine study to stage their disease. After the disease is staged, a 6-month study is performed to determine whether the tumor is fast growing or slow growing.
CN V, or trigeminal nerve, schwannomas are the most common type seen after vestibular schwannomas. Oculomotor schwannomas are the third most common schwannomas seen intracranially. Occasionally, it is difficult to distinguish whether these schwannomas have arisen from the oculomotor, trochlear, or abducent nerve, especially when the tumor arises within the cavernous sinus. Trochlear and abducent schwannomas are extremely rare, with only a handful of cases reported in the literature.
Facial nerve schwannomas may also be seen, although these are difficult to distinguish radiographically from vestibular schwannomas. Some patients present with small facial schwannomas that are encountered with large tumors in which the distinction between the facial nerve and cochlear vestibular nerve cannot be found. CN III and V schwannomas are usually slow growing and require treatment only when significant growth has occurred, or other intracranial complications are imminent.
Lower CN schwannomas can be significant in NF2 because these can lead to speech and swallowing disorders. When bilateral lower CN schwannomas or jugular foramen meningiomas are associated with these tumors, the patient may develop aspiration problems, which can cause significant morbidity. Glossopharyngeal, vagal, and hypoglossal neuropathies resulting from schwannomas on CN IX, X, and XII may lead to speech and swallowing disorders. Glossopharyngeal schwannomas are the most common schwannomas of the jugular foramen and may manifest with swallowing difficulty, which may lead to the requirement of gastrostomy feeding tubes for nutritional status.
Vagal nerve defects may contribute to swallowing difficulties related to esophageal dysmotility. Vagal nerve deficits may manifest with voice hoarseness owing to vocal cord paralysis, but the most imminent issue is aspiration, which occurs because of loss of sensory innervation to the larynx and loss of the reflexive airway protective mechanisms. Aspiration is often silent in such cases and leads to life-threatening pulmonary complications, including pneumonia. Tracheotomy may be required, leading to other potential life-threatening complications, including pulmonary infection. It is thought that lower CN neuropathies may contribute to mortality associated with NF2.
Hearing Changes in Patients with Vestibular Schwannomas
Hearing loss is well documented as the most common presenting symptom in patients who have vestibular schwannomas. Auditory changes over time in patients with vestibular schwannoma are less well known. Rosenberg studied the natural history of 80 patients with non-NF2 unilateral vestibular schwannomas for an average of 4.4 years and found a positive correlation between tumor growth and worsening pure tone average. However, there was no statistically significant correlation between positive tumor growth and speech discrimination, change in ABR, and bithermal caloric electronystagmography testing result over time.
Lalwani and colleagues reported that pure tone pattern speech receptive thresholds and word recognition scores were significantly worse in patients with NF2 who had a mild form of NF2 and large tumors compared with patients with mild NF2 with small tumors. Loss of acoustic reflexes and prolonged wave III and V were also associated with larger tumors. In contrast, patients with severe NF2 showed no relationship among tumor size and pure tone levels, speech discrimination thresholds, or word recognition scores. The lack of association may have been caused by complete loss of hearing in patients with severe NF2 at the time of the assessment. The larger tumors were also associated with prolonged ABR waves III and V latencies. No data across time were reported. In general, hearing is progressively impaired with increasing growth of vestibular schwannomas necessitating surgical intervention or medical treatments in patients with NF2.
The natural history study evaluated hearing changes prospectively in 63 patients newly diagnosed with NF, and found that hearing was stable after diagnosis. During the first 2 years after diagnosis, 27% of patients had a significant change in their hearing, and 73% had very stable hearing during the 2-year period. Patients with a family history of NF2 had more stable hearing after an initial diagnosis compared with patients without a family history. Patients with a family history are usually diagnosed at a younger age; the stability of hearing in this group may represent the younger age group. The better the hearing in the newly diagnosed patients, the more stable the hearing. Hearing changes did not vary between ears, with each ear acting independently.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


