A comprehensive discussion of neurofibromatosis 2 (NF2) is presented, including clinical characteristics, symptoms, diagnosis, tumor types, prevalence and incidence, genetic testing, imaging, treatment options, and follow-up management of NF2.
Neurofibromatosis 2 (NF2) is a rare syndrome characterized by:
- •
Bilateral vestibular schwannomas
- •
Multiple meningiomas
- •
Cranial nerve tumors
- •
Spinal tumors
- •
Eye abnormalities.
NF2 presents unique challenges to the otologists because hearing loss may be the presenting complaint leading to the diagnosis of the disorder. NF2 is invasive, requiring a multispecialist team approach for the evaluation and treatment of the disorder. The primary impairment is hearing loss resulting from bilateral vestibular schwannomas. NF2 must be differentiated from neurofibromatosis 1 (NF1); although the names are linked, the disease entities are distinctly different. This article reviews the clinical characteristics of NF2 and the current recommendations for evaluation and treatment.
NF2 differentiated from NF1
NF1 has distinctly different characteristics from NF2. NF1 and NF2 have been differentiated as completely different genetic diseases based on the chromosome responsible for the disease. NF1 has been localized to chromosome 17 and NF2 to chromosome 22.
NF1 is a multisystem disorder in which some features may be present at birth and others are age-related manifestations. A National Institutes of Health (NIH) Consensus Development Conference identified the following 7 features of the disease, of which 2 or more are required to establish the diagnosis of NF1:
- 1.
Six café au lait spots of 5 mm or more in longest diameter in prepubertal patients and 15 mm in the longest diameter in postpubertal patients.
- 2.
Two or more neurofibromas of any type or 1 plexiform neurofibroma.
- 3.
Freckling in the axilla or inguinal regions.
- 4.
Optic glioma (optic pathway glioma).
- 5.
Two or more Lisch nodules (iris hamartomas).
- 6.
Distinct osseous lesions, such as sphenoid wing dysplasia or cortical thinning of the cortex of long bones with or without pseudoarthrosis.
- 7.
First-degree relative (parent, sibling, or child) with NF1 according to the above-listed criteria.
Some patients also manifest learning disabilities or language disorders. A careful examination and detailed history of the patient’s symptoms help distinguish NF1 and NF2.
Clinical characteristics of NF2
Definition
The NIH Consensus Development Conference also developed guidelines for the diagnosis of NF2. NF2 is distinguished by bilateral vestibular schwannomas with multiple meningiomas, cranial nerve tumors, optic gliomas, and spinal tumors. A definite diagnosis is made based on the presence of bilateral vestibular schwannomas or developing a unilateral vestibular schwannoma by 30 years and a first-degree blood relative with NF2, or the presence of a unilateral vestibular schwannoma and developing at least 2 of the following conditions known to be associated with NF2: meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacity/juvenile cortical cataract ( Box 1 ).
Confirmed (definite) NF2
Bilateral VS or family history of NF2 (first-degree family relative) plus
- 1.
Unilateral VS before 30 years of age or
- 2.
Any 2 of the following: meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
- 1.
Presumptive or probable NF2: Should evaluate for NF2
Unilateral VS before 30 years of age plus at least 1 of the following: meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
Multiple meningiomas (≥2) plus unilateral VS before 30 years of age or 1 of the following: glioma, schwannoma, or juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
Abbreviation: VS, vestibular schwannoma.
NF2 Clinical Presentation
There may be significant heterogeneity in the presentation of the disease from one individual to the other. Some individuals may have a very mild form of the disease, such as small vestibular schwannomas manifesting in an older individual, whereas some children present with multiple intracranial tumors at a very young age. Despite the heterogeneity of the disease, the expression of NF2 tends to be very similar within a family.
There is a significant genetic component to the disease with much variability within the parameters of the observed phenotype. Studies have shown that a truncating mutation (nonsense and frame shift) may be linked with a more severe form of NF2. The more severe form of NF2 is termed the Wishart form. Individuals with this severe form present with an early onset of the disease with multiple intracranial schwannomas and meningiomas that result in blindness, deafness, paralysis, and possible death by 40 years. Despite the strong genotype-phenotype correlation, individual differences in tumor growth occur within individuals, making it difficult to predict how an individual’s tumors will change over time, even when the genotype is known. The milder form of NF2, or the Gardner form, is less debilitating. Schwannomas may remain stable for many years; few meningiomas develop; and patients may not develop symptoms until later in life and often have fewer associated disabilities. The genetic basis of the mild form has not been well characterized. Many of these may be mosaic forms of the disease.
Prevalence and Incidence of NF2
The average age of diagnosis of NF2 is 25 years; however, many patients present with symptoms before the diagnosis. There is an average delay in the diagnosis of approximately 7 years. There is no difference in the proportion of men versus women who develop NF2, and no prevalence has been described based on ethnicity. Epidemiologic studies place the incidence of NF2 between 1 in 33,000 live births and 1 in 87,410 live births.
Imaging Studies for NF2
All patients suspected of NF2 should have a high-quality magnetic resonance imaging (MRI) scan performed with thin cuts through the internal auditory canal (IAC). All patients diagnosed with a unilateral vestibular schwannoma should have a dedicated IAC series performed to ensure that there is no tumor on the opposite side. Patients diagnosed with NF2 should have a complete spine series performed to evaluate the spine and stage the disease. Commonly, small spinal tumors may be found in the cauda equina, and occasionally, a large asymptomatic schwannoma or meningioma may be found in the spine that could require treatment. Early treatment of spinal tumors can significantly reduce the morbidity associated with these tumors. Older patients who present with bilateral IAC tumors must be worked up for other carcinomas. It is unusual for patients older than 40 years to present with NF2, although with more sensitive MRI scanning techniques, most of these individuals are being diagnosed at an older age. Metastasis may rarely manifest with bilateral IAC lesions, and carcinoma should be ruled out in any patient older than 40 years who presents with bilateral IAC lesions.
A patient diagnosed with NF2 should have a complete cranial MRI scan with cervical, thoracic, and lumbar spinal imaging, which serves as a baseline. A 6-month follow-up MRI scan is recommended for patients with intracranial tumors. If the tumors exhibit stability, a yearly MRI scan of the intracranial structures is performed. Large tumors in the spine may be monitored with a similar frequency. If no spinal tumors are present, spinal imaging should be performed when the patient becomes symptomatic. Monitoring small-to-medium spinal tumors every 3 years is recommended when these tumors are present. Intracranial imaging is performed on a yearly basis unless studies over several years indicate stability of the tumors.
| Patient’s Status | Imaging Study |
| Unilateral vestibular schwannoma | IAC |
| Screening for NF2 | IAC |
| Diagnosis of NF2 | AC/Cranial Complete Spine Series ✓ Initial 6 month follow-up ✓ 12 month scan if stable ✓ 3 years for small spine tumors |
Molecular Genetics of NF2
The NF2 gene was mapped to chromosome 22q12.2 in 1993. The NF2 gene located at chromosome 22 codes for a tumor suppressor protein termed Merlin or Schwannomin, which negatively regulates Schwann cell production. The loss of this protein allows overproduction of Schwann cells. The mutation in the NF2 chain predisposes individuals to developing a schwannoma when the second hit occurs to the gene; control of Schwann cells is lost or mutated within the cell. Various types of mutations have been identified, including single-base substitutions, insertions, and deletions. The mild, or Gardner, type of NF2 may be associated with missense mutations, whereas associations between the other mutations and phenotypes are not as clear. The occurrence of NF2 is not restricted to families known to carry the mutation. Frequently, genetic mosaicism occurs, which may not be detected by common mutation analysis techniques. Unilateral vestibular schwannomas may exhibit the same type of genetic markers as NF2. The mutations in unilateral vestibular schwannomas are confined to the affected tumor tissue. In patients with NF2, the mutation is present in all cell types.
Family History of NF2
NF2 is an autosomal dominant disease, and 50% of children of the affected individuals are at risk for developing the disease. Of the patients diagnosed with NF2, 50% present with a family history of NF2 and the other half have no family history of NF2 and are considered founder cases. The presentation and phenotype of NF2 tend to be similar within families. The likelihood of NF2 occurring in related individuals who do not exhibit clinical symptoms similar to those in an affected family member is small. Consideration must still be given to screening these individuals for risks, despite the lack of clinical symptoms. Individuals at risk for developing NF2, including children of NF2-affected patients and their siblings, must be screened to provide an early diagnosis. Fifty percent of all children of patients with NF2 are found to have the disease. Siblings of a patient diagnosed with NF2 are at risk, especially if the parent also has NF2. The type of screening and timing of screening depend on the NF2 center’s preference. Early screening is advocated so that tumors may be diagnosed presymptomatically. MRI or genetic blood testing may be used for screening.
Screening for NF2
MRI
MRI screening of potentially affected individuals uses a postcontrast T1-weighted sequence of the full head with thin cuts through the IAC. A dedicated IAC MRI scan identifies most patients with NF2 by showing vestibular schwannomas. Screening of the spine and ophthalmologic examination should be considered if the cranial MRI scan result is positive. An audiogram (pure tone thresholds) or a current clinical standard auditory brainstem response (ABR) testing is likely to miss small vestibular schwannomas. MRI can diagnose presymptomatic lesions.
MRI is recommended for at-risk children when this test can be performed without sedation; this usually can be done when the child is 7 to 9 years old. A recommended first step for children younger than 7 years is an audiogram. Any child with an NF2-associated symptom, such as hearing loss or facial weakness, should be screened irrespective of the need for sedation or age; MRI should be performed as soon as possible after the symptoms become apparent.
Molecular testing
Identification of the NF2 gene and chromosome 22 has made genetic testing possible. Patients with NF2 should see a genetic counselor to discuss the hereditary consequences of the disease. Blood testing for the mutation identifies the defect of the NF2 gene in approximately 70% to 75% of patients with a known diagnosis of NF2. Blood screening of family members is done depending on whether the defect is identified in the affected individual or not. The use of blood screening for patients without a diagnosis of NF2 or with a suspected diagnosis of NF2 is not recommended. New mutations in patients with mild presentation are most likely to be missense mutations and are difficult to identify with genetic testing of patients with NF2. The best molecular testing is performed on fresh vestibular schwannoma tissue. Any patient with a planned vestibular schwannoma removal should be offered the opportunity to have their tumor tissue tested for the genetic mutation. This is the most sensitive test and allows the identification of the genetic mutation in most patients. The results of this test can then be used to screen relatives for NF2. There are currently 2 clinical laboratories that perform this test (the Massachusetts General Hospital, Boston, MA, and the University of Alabama, Tuscaloosa, AL) and many research laboratories associated with NF centers that may perform the test on recently resected tissue. The tissue must be kept frozen, and testing should be coordinated with the laboratories before surgical resection.
Clinical characteristics of NF2
Definition
The NIH Consensus Development Conference also developed guidelines for the diagnosis of NF2. NF2 is distinguished by bilateral vestibular schwannomas with multiple meningiomas, cranial nerve tumors, optic gliomas, and spinal tumors. A definite diagnosis is made based on the presence of bilateral vestibular schwannomas or developing a unilateral vestibular schwannoma by 30 years and a first-degree blood relative with NF2, or the presence of a unilateral vestibular schwannoma and developing at least 2 of the following conditions known to be associated with NF2: meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacity/juvenile cortical cataract ( Box 1 ).
Confirmed (definite) NF2
Bilateral VS or family history of NF2 (first-degree family relative) plus
- 1.
Unilateral VS before 30 years of age or
- 2.
Any 2 of the following: meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
- 1.
Presumptive or probable NF2: Should evaluate for NF2
Unilateral VS before 30 years of age plus at least 1 of the following: meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
Multiple meningiomas (≥2) plus unilateral VS before 30 years of age or 1 of the following: glioma, schwannoma, or juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract
Abbreviation: VS, vestibular schwannoma.
NF2 Clinical Presentation
There may be significant heterogeneity in the presentation of the disease from one individual to the other. Some individuals may have a very mild form of the disease, such as small vestibular schwannomas manifesting in an older individual, whereas some children present with multiple intracranial tumors at a very young age. Despite the heterogeneity of the disease, the expression of NF2 tends to be very similar within a family.
There is a significant genetic component to the disease with much variability within the parameters of the observed phenotype. Studies have shown that a truncating mutation (nonsense and frame shift) may be linked with a more severe form of NF2. The more severe form of NF2 is termed the Wishart form. Individuals with this severe form present with an early onset of the disease with multiple intracranial schwannomas and meningiomas that result in blindness, deafness, paralysis, and possible death by 40 years. Despite the strong genotype-phenotype correlation, individual differences in tumor growth occur within individuals, making it difficult to predict how an individual’s tumors will change over time, even when the genotype is known. The milder form of NF2, or the Gardner form, is less debilitating. Schwannomas may remain stable for many years; few meningiomas develop; and patients may not develop symptoms until later in life and often have fewer associated disabilities. The genetic basis of the mild form has not been well characterized. Many of these may be mosaic forms of the disease.
Prevalence and Incidence of NF2
The average age of diagnosis of NF2 is 25 years; however, many patients present with symptoms before the diagnosis. There is an average delay in the diagnosis of approximately 7 years. There is no difference in the proportion of men versus women who develop NF2, and no prevalence has been described based on ethnicity. Epidemiologic studies place the incidence of NF2 between 1 in 33,000 live births and 1 in 87,410 live births.
Imaging Studies for NF2
All patients suspected of NF2 should have a high-quality magnetic resonance imaging (MRI) scan performed with thin cuts through the internal auditory canal (IAC). All patients diagnosed with a unilateral vestibular schwannoma should have a dedicated IAC series performed to ensure that there is no tumor on the opposite side. Patients diagnosed with NF2 should have a complete spine series performed to evaluate the spine and stage the disease. Commonly, small spinal tumors may be found in the cauda equina, and occasionally, a large asymptomatic schwannoma or meningioma may be found in the spine that could require treatment. Early treatment of spinal tumors can significantly reduce the morbidity associated with these tumors. Older patients who present with bilateral IAC tumors must be worked up for other carcinomas. It is unusual for patients older than 40 years to present with NF2, although with more sensitive MRI scanning techniques, most of these individuals are being diagnosed at an older age. Metastasis may rarely manifest with bilateral IAC lesions, and carcinoma should be ruled out in any patient older than 40 years who presents with bilateral IAC lesions.
A patient diagnosed with NF2 should have a complete cranial MRI scan with cervical, thoracic, and lumbar spinal imaging, which serves as a baseline. A 6-month follow-up MRI scan is recommended for patients with intracranial tumors. If the tumors exhibit stability, a yearly MRI scan of the intracranial structures is performed. Large tumors in the spine may be monitored with a similar frequency. If no spinal tumors are present, spinal imaging should be performed when the patient becomes symptomatic. Monitoring small-to-medium spinal tumors every 3 years is recommended when these tumors are present. Intracranial imaging is performed on a yearly basis unless studies over several years indicate stability of the tumors.
| Patient’s Status | Imaging Study |
| Unilateral vestibular schwannoma | IAC |
| Screening for NF2 | IAC |
| Diagnosis of NF2 | AC/Cranial Complete Spine Series ✓ Initial 6 month follow-up ✓ 12 month scan if stable ✓ 3 years for small spine tumors |
Molecular Genetics of NF2
The NF2 gene was mapped to chromosome 22q12.2 in 1993. The NF2 gene located at chromosome 22 codes for a tumor suppressor protein termed Merlin or Schwannomin, which negatively regulates Schwann cell production. The loss of this protein allows overproduction of Schwann cells. The mutation in the NF2 chain predisposes individuals to developing a schwannoma when the second hit occurs to the gene; control of Schwann cells is lost or mutated within the cell. Various types of mutations have been identified, including single-base substitutions, insertions, and deletions. The mild, or Gardner, type of NF2 may be associated with missense mutations, whereas associations between the other mutations and phenotypes are not as clear. The occurrence of NF2 is not restricted to families known to carry the mutation. Frequently, genetic mosaicism occurs, which may not be detected by common mutation analysis techniques. Unilateral vestibular schwannomas may exhibit the same type of genetic markers as NF2. The mutations in unilateral vestibular schwannomas are confined to the affected tumor tissue. In patients with NF2, the mutation is present in all cell types.
Family History of NF2
NF2 is an autosomal dominant disease, and 50% of children of the affected individuals are at risk for developing the disease. Of the patients diagnosed with NF2, 50% present with a family history of NF2 and the other half have no family history of NF2 and are considered founder cases. The presentation and phenotype of NF2 tend to be similar within families. The likelihood of NF2 occurring in related individuals who do not exhibit clinical symptoms similar to those in an affected family member is small. Consideration must still be given to screening these individuals for risks, despite the lack of clinical symptoms. Individuals at risk for developing NF2, including children of NF2-affected patients and their siblings, must be screened to provide an early diagnosis. Fifty percent of all children of patients with NF2 are found to have the disease. Siblings of a patient diagnosed with NF2 are at risk, especially if the parent also has NF2. The type of screening and timing of screening depend on the NF2 center’s preference. Early screening is advocated so that tumors may be diagnosed presymptomatically. MRI or genetic blood testing may be used for screening.
Screening for NF2
MRI
MRI screening of potentially affected individuals uses a postcontrast T1-weighted sequence of the full head with thin cuts through the IAC. A dedicated IAC MRI scan identifies most patients with NF2 by showing vestibular schwannomas. Screening of the spine and ophthalmologic examination should be considered if the cranial MRI scan result is positive. An audiogram (pure tone thresholds) or a current clinical standard auditory brainstem response (ABR) testing is likely to miss small vestibular schwannomas. MRI can diagnose presymptomatic lesions.
MRI is recommended for at-risk children when this test can be performed without sedation; this usually can be done when the child is 7 to 9 years old. A recommended first step for children younger than 7 years is an audiogram. Any child with an NF2-associated symptom, such as hearing loss or facial weakness, should be screened irrespective of the need for sedation or age; MRI should be performed as soon as possible after the symptoms become apparent.
Molecular testing
Identification of the NF2 gene and chromosome 22 has made genetic testing possible. Patients with NF2 should see a genetic counselor to discuss the hereditary consequences of the disease. Blood testing for the mutation identifies the defect of the NF2 gene in approximately 70% to 75% of patients with a known diagnosis of NF2. Blood screening of family members is done depending on whether the defect is identified in the affected individual or not. The use of blood screening for patients without a diagnosis of NF2 or with a suspected diagnosis of NF2 is not recommended. New mutations in patients with mild presentation are most likely to be missense mutations and are difficult to identify with genetic testing of patients with NF2. The best molecular testing is performed on fresh vestibular schwannoma tissue. Any patient with a planned vestibular schwannoma removal should be offered the opportunity to have their tumor tissue tested for the genetic mutation. This is the most sensitive test and allows the identification of the genetic mutation in most patients. The results of this test can then be used to screen relatives for NF2. There are currently 2 clinical laboratories that perform this test (the Massachusetts General Hospital, Boston, MA, and the University of Alabama, Tuscaloosa, AL) and many research laboratories associated with NF centers that may perform the test on recently resected tissue. The tissue must be kept frozen, and testing should be coordinated with the laboratories before surgical resection.
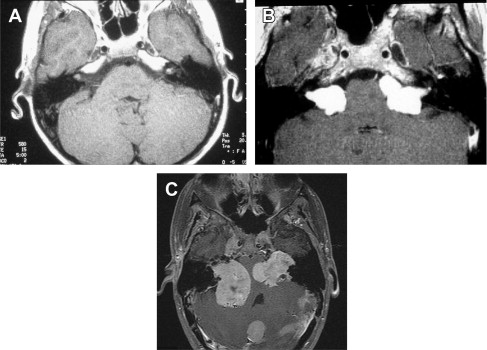
Schwannoma tumor types
Bilateral vestibular schwannomas (acoustic neuromas) are benign neoplasms of the acoustic or eighth cranial nerve ( Fig. 1 ). The tumors are thought to arise from the glioma: Schwann cell junction within the internal auditory meatus. The tumors most commonly arise from the superior vestibular nerve, although with NF2, tumors may be found on the cochlear and facial nerves within the internal auditory meatus. The consequences of a vestibular schwannoma are numerous, including hearing loss progressing to deafness, dizziness and balance problems, tinnitus, facial nerve paralysis, brainstem compression, and, if left untreated, death.