Myopathies of Neuro-Ophthalmologic Significance
In this chapter, we consider disorders that produce ocular motor dysfunction from involvement of the extraocular muscles. Some of these disorders affect only the extraocular muscles, whereas others are multisystem disorders in which various organ systems are also affected.
Developmental Disorders of Extraocular Muscle
Anomalous development of extraocular muscles probably is more common than the literature would indicate. Many of these anomalies are recognized only during surgical procedures or at autopsy. The most common congenital anomalies of the extraocular muscles are agenesis, anomalous insertions or origins, and the adherence and fibrosis syndromes.
Agenesis of the Extraocular Muscles
Most cases of agenesis of the extraocular muscles involve only a single muscle. Isolated agenesis of the lateral rectus muscle, medial rectus muscle, inferior rectus muscle, superior rectus muscle, and superior oblique muscle are all well described, particularly in children with craniostenosis.
Anomalies of Extraocular Muscle Origin, Insertion, and Structure
Abnormal origins of extraocular muscles are rare. Somewhat more commonly responsible for ocular motor dysfunction is an abnormal insertion of an extraocular muscle. Abnormal insertions of extraocular muscles, like agenesis of the muscles, often occur in children with craniostenosis.
Occasionally, an extraocular muscle shows underaction because of an abnormally increased length. In other instances, an anomalous muscle slip or fibrous band may be present. This phenomenon appears to be responsible for most of the congenital cases of superior oblique tendon dysfunction called Brown syndrome. Patients with this syndrome show absence of elevation in adduction, improvement of elevation in the primary position, and normal or near-normal elevation in abduction (Fig. 20.1). Forced duction testing (see Chapter 16) reveals mechanical limitation of motion upward and inward in the involved eye, but upward saccadic velocities are normal.
Congenital Adherence and Fibrosis Syndromes
Congenital fibrosis of the extraocular muscles (CFEOM) is the term used to describe several hereditary disorders characterized by (a) ophthalmoplegia, (b) ptosis, (c) fibrosis of all the extraocular muscles, (d) fibrosis of Tenon capsule, (e) adhesions between muscles, Tenon capsule, and globe, and (f) inelasticity and fragility of the conjunctiva (Fig. 20.2). CFEOM is associated with mutations at three distinct loci, FEOM1, FEOM2, and FEOM3, located on chromosomes 12, 11, and 16, respectively. FEOM1 and FEOM3 mutations produce autosomal-dominant CFEOM, whereas the FEOM2 mutation produces autosomal-recessive CFEOM. Most patients with these disorders have only ocular motor dysfunction, but some patients have facial diparesis, and individual patients have been described who also have inguinal hernias and cryptorchidism or a cleft palate.
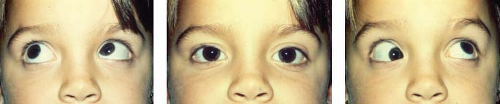 Figure 20.1 Congenital Brown syndrome in an 8-year-old girl. Note the patient’s inability to elevate the right eye in adduction. (Courtesy of Dr. Michael X. Repka.) |
Although histopathologic findings in some patients with CFEOM include replacement of muscle tissue with fibrous tissue and degenerative changes in the muscles, these findings are secondary to maldevelopment of the oculomotor (III) nerve, trochlear (IV) nerve, or both. Thus, CFEOM is actually a disorder similar in origin to Duane and Möbius syndromes.
Congenital Myopathies
Two types of congenital, primarily nonprogressive myopathies can affect the extraocular and eyelid muscles, producing bilateral ptosis, ophthalmoparesis, and facial weakness (Fig. 20.3). One group consists of disorders that are similar to one another clinically but have distinctive histopathologic features. They are called collectively structurally defined congenital myopathies and include: (1) central core myopathy, (2) nemaline myopathy, (3) centronuclear (or myotubular) myopathy, (4) multicore disease, and (5) congenital fiber-type disproportion.
A second group of congenital myopathies remains to be characterized fully. These disorders generally present with congenital hypotonia or with early-onset generalized weakness that does not progress. Some have ocular features; others do not. Skeletal deformities, such as high-arched palate, scoliosis, and a slender body habitus may be present.
Most congenital myopathies are mild and nonprogressive or slowly progressive. Some cases, however, are severe, relentlessly progressive, or both, resulting in substantial debilitation.
 Figure 20.2 Congenital fibrosis syndrome in a brother and sister. Because of the bilateral ptosis and ophthalmoplegia, both children have adopted a head posture with the chin elevated. (Courtesy of Dr. Stewart M. Wolff.) |
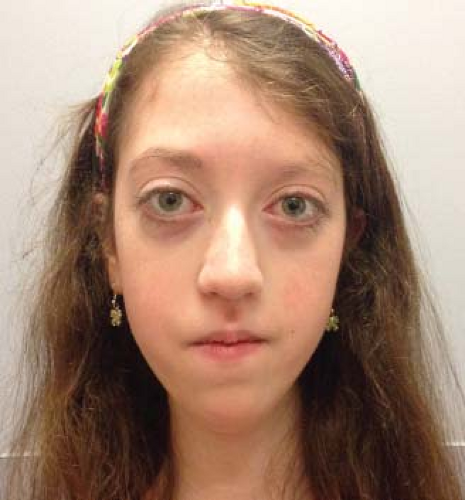 Figure 20.3 Appearance of a 14-year-old girl with severe congenital fiber-type disproportion. Note bilateral asymmetric ptosis, inferior scleral show due to laxity of lower lids, and flaccid facies. The patient also has orbital dystopia. (Courtesy of Dr. Thomas Crawford.) |
Congenital myopathies must be differentiated from other causes of congenital hypotonic weakness such as various forms of spinal muscular atrophy. This distinction usually is fairly simple, however, as most of these latter conditions are not associated with either ptosis or ophthalmoparesis/plegia. Thus, an infant with congenital hypotonia associated with ptosis, external ophthalmoplegia, or both almost always has a myopathy. Nevertheless, conditions other than congenital myopathies, including neonatal myasthenia gravis and infant botulism, also can cause hypotonia, weakness, facial paresis, and ophthalmoparesis.
Muscular Dystrophies
The term “muscular dystrophy” covers a group of genetically determined disorders that cause progressive weakness and wasting of the skeletal muscles and that are assumed to affect the muscle cell directly. Some forms cause death after 15 to 20 years, whereas others are compatible with a normal life expectancy. The distinction between the terms “myopathy” and “dystrophy” breaks down, however, when it is realized that some glycogenolytic myopathies, like acid maltase deficiency, can progress inexorably like dystrophies, and some dystrophies, like Becker dystrophy, may hardly progress at all. Nevertheless, the diagnostic category is a tradition. Although the diagnosis of this group of disorders cannot be established by morphology alone, histologic examination of an affected muscle can exclude other myopathies (e.g., the congenital myopathies and the mitochondrial myopathies). In others, molecular genetic testing can provide the correct diagnosis.
Muscular dystrophies are historically subdivided on the basis of age of onset, mode of inheritance, and clinical features as follows:
The dystrophin-deficient dystrophies, including the severe X-linked pseudohypertrophic muscular dystrophy of Duchenne and its more benign “Becker” variant
The dystrophin-associated glycoprotein disorders that may resemble Duchenne or Becker dystrophy, including mutations of merosin and the α-, β-, γ-, or δ-sarcoglycans
Various limb-girdle dystrophies, including severe childhood-onset autosomal-recessive (chromosome 13) muscular dystrophy, juvenile scapulohumeral dystrophy of Erb, pelvifemoral dystrophy of Leyden and Möbius, and adult-onset autosomal-dominant limb-girdle dystrophy
The facioscapulohumeral dystrophy of Landouzy and Déjérine
Distal dystrophies (myopathies) of Welander, Miyoshi, and others
Congenital muscular dystrophies (CMDs), including Fukuyama dystrophy, muscle–eye–brain disease, and Walker–Warburg syndrome
Myotonic muscular dystrophy
Proximal myotonic myopathy (PROMM)
Oculopharyngeal muscular dystrophy (OPMD)
Only the last four of these disorders are of neuro-ophthalmologic importance and are discussed below.
Congenital Muscular Dystrophies
Patients with dystrophic muscle pathology associated with symptoms present at birth and a variable clinical course are said to have CMD. These individuals have generalized muscle atrophy and weakness that is symmetric and predominates in proximal muscles. In addition, nonmuscular ocular involvement is common in three congenital dystrophy syndromes that are really multisystemic disorders of eye, brain, and skeletal muscle. These three—the Fukuyama type of CMD, so-called muscle–eye–brain disease, and the Walker–Warburg syndrome—are associated with mutations that result in protein glycosylation defects.
Fukuyama Congenital Muscular Dystrophy
The Fukuyama type of CMD is transmitted as an autosomal-recessive trait with no gender predilection, a high incidence of consanguinity, and a high frequency of affected siblings. The mutant gene, located on chromosome 9q31, encodes fukutin, a putative glycosyltransferase.
Patients with Fukuyama dystrophy have CMD associated with central nervous system (CNS) involvement, including mental retardation and convulsions. Children are severely weak from birth, and only a few children can walk without assistance. Most survive beyond infancy to remain relatively stable, but the average life span is 8 to 10 years. Serum levels of creatinine kinase (CK) are elevated, and the muscle biopsy shows myopathic and fibrotic changes. Ventricular enlargement and hypoplasia of the cerebellar vermis are seen with computed tomographic (CT) scanning, and magnetic resonance (MR) imaging shows severe signal abnormalities in white matter.
The neuropathologic findings in Fukuyama dystrophy include polymicrogyria of the cerebrum and cerebellum, loss of cerebral cytoarchitecture with demyelination and proliferation of interstitial tissue, occasionally absent or hypoplastic corpus callosum, and brain stem pyramidal tract abnormalities. Chronic meningeal thickening, lymphocytic infiltration of intracranial tissues, and proliferation of meningeal tissue and blood vessels are less common.
Ocular involvement in Fukuyama CMD is inconsistent, but it is milder than that in muscle–eye–brain
disease and Walker–Warburg syndrome (see below). Reported findings include occasional optic nerve hypoplasia and optic atrophy, as well as high myopia, cataracts, weakness of the orbicularis oculi muscles, and retinal abnormalities that may be mild or severe, including mottling of the retinal pigment epithelium, abnormal retinal vessels, incomplete retinal vascularization with temporally displaced foveae, and dysplasia.
disease and Walker–Warburg syndrome (see below). Reported findings include occasional optic nerve hypoplasia and optic atrophy, as well as high myopia, cataracts, weakness of the orbicularis oculi muscles, and retinal abnormalities that may be mild or severe, including mottling of the retinal pigment epithelium, abnormal retinal vessels, incomplete retinal vascularization with temporally displaced foveae, and dysplasia.
Muscle–Eye–Brain Disease
This autosomal-recessive disorder is associated with homozygous mutations in O-mannose beta-1,2–N-acetylglucosaminyltransferase (POMGnT1), an enzyme that participates in O-mannosyl glycan synthesis. It is characterized by severe congenital hypotonic weakness from both progressive CNS disease and CMD. Although the cerebral and visual abnormalities are severe, most patients reach adulthood (average age of death, 18 years). As in Fukuyama dystrophy, levels of serum CK are elevated, and muscle biopsy shows mild myopathy with fibrosis.
Ocular abnormalities are more consistent and severe in muscle–eye–brain disease than in Fukuyama dystrophy, but they are less uniform than in Walker–Warburg syndrome. High myopia is common. Other ocular findings include severe generalized loss of retinal ganglion cells, retinochoroidal scars, a pronounced preretinal membrane and gliosis, mottled retinal pigment epithelium, and mild optic nerve and chiasm atrophy with reactive gliosis. The microscopic and macroscopic features of the cortex in these cases tend to be highly abnormal, showing argyric areas, coarse and nodular gyri, or total disorganization. The cerebellar vermis and brainstem may be hypoplastic.
Walker–Warburg Syndrome
The Walker–Warburg syndrome is an autosomal-recessive disorder with more severe findings than either Fukuyama dystrophy or muscle–eye–brain disease. It is caused by homozygous mutations in the gene encoding O-mannosyltransferase 1 (POMT1) that, like POMGnT1 (the mutant protein in muscle–eye–brain disease), participates in O-mannosyl glycan synthesis. The neuropathologic features of Walker–Warburg syndrome are the same as those of muscle–eye–brain disease, but they are more extensive and include the macroscopic features of arrhinencephaly: cephalocele, hemispheric fusion, agenesis of the corpus callosum, or a combination of these manifestations.
The ocular malformations in Walker–Warburg syndrome, like the neuropathologic features, are more severe than in muscle–eye–brain disease. Typical ocular features include retinal dysplasia and nonattachment, persistent hyperplastic primary vitreous, optic nerve atrophy, microphthalmia, corneal opacities, congenital cataract, and congenital glaucoma.
The skeletal muscle biopsy changes in Walker–Warburg syndrome are similar to those of muscle–eye–brain disease and less severe than those in Fukuyama dystrophy. Levels of serum CK are elevated in this disorder as it is in the other two.
Myotonic Muscular Dystrophies
Myotonic dystrophy (dystrophia myotonica 1 [DM1]) and the less severe PROMM (DM2) are autosomal-dominant disorders associated with nucleotide repeat expansions in untranslated regions of the respective target genes. The genetic defect in DM1 is amplification of an unstable trinucleotide CTG repeat located in the 3′ untranslated region of the gene that encodes dystrophia myotonica protein kinase (DMPK) on chromosome 19. DM1 is highly variable in severity and age of onset, and it exhibits “anticipation,” the phenomenon of increasing severity of inherited disease in successive generations of an affected family. The number of CTG repeats ranges from about 50 in patients who are mildly affected to thousands in severely affected patients. Amplification of the CTG repeat is the molecular basis for genetic anticipation. Although the mutation does not alter the protein-coding region of the DMPK gene, the CTG repeats can disrupt RNA splicing and cellular metabolism by interacting with RNA-binding proteins.
PROMM/DM2 is caused by a CCTG expansion (approximately 5,000 repeats) in intron 1 of the gene encoding zinc finger protein 9 (ZNF9). Like the CTG expansion of the DMPK gene in DM1, the CCTG expansion does not alter the protein-coding region of the ZNF9 gene. However, the CCTG repeats can disrupt RNA splicing and cellular metabolism by interacting with RNA-binding proteins.
Myotonic Dystrophy
Myotonic dystrophy (DM1) is the most common inherited myopathy in adults, with a prevalence of approximately 5 per 100,000 and men and women being equally affected. It is a multisystem disorder characterized by progressive wasting and weakness of distal muscles and myotonia.
Systemic and Ocular Manifestations
Many patients can be recognized instantly by their characteristic frontal balding, long face, ptosis, hollowing of the masseter and temporalis muscles, slackened mouth, facial weakness, and thin neck and limbs (Fig. 20.4). Other features include intellectual impairment, testicular atrophy, excessive daytime somnolence, insulin resistance, and cardiac conduction defects. The most important finding on physical examination is myotonia—involuntary delayed relaxation following a contraction, such as after sustained handgrip; however, most patients with DM1 complain of weakness, not myotonia.
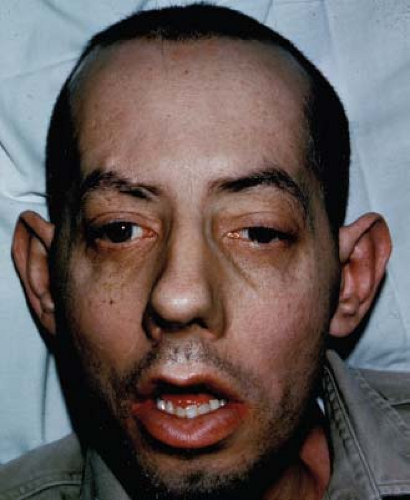 Figure 20.4 Clinical appearance of a 56-year-old man with myotonic dystrophy (DM1). Note bilateral ptosis, exotropia, myopathic facies, and frontal baldness typical of this disorder. |
Cataract is the most common ocular abnormality in patients with DM1, occurring in nearly 100% of patients with the disease. The severity of the cataract is not related to the severity of the disease. Myotonic cataracts may be of two types. The first consists of tiny opaque, white, or—most often—iridescent, colored (usually red, green, or blue) crystals that are located in a thin band of anterior and posterior cortex just beneath the lens capsule (Fig. 20.5A). The second type is a stellate grouping of opacities at the posterior pole along the posterior suture lines, producing a spoke-like appearance (Fig. 20.5B).
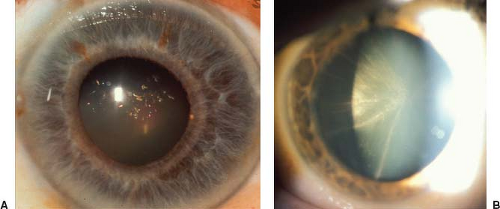 Figure 20.5 Typical appearance of cataracts in myotonic dystrophy (DM1). A: Numerous dot opacities, each having a different color, located primarily in the posterior subcortical region of the lens. B: Spoke-like opacities of the posterior subcortical region of the lens. |
In addition to cataracts, ptosis frequently is present in patients with DM1 and may be mild (Fig. 20.4) or profound (Fig. 20.6A




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
