HLA-B27-associated uveitis is recurrent with alternating bilateral involvement.
Cells and flare in the anterior chamber
Circumcorneal ciliary flush
Nongranulomatous: fine keratic precipitates (KPs) on corneal endothelium
Granulomatous: “mutton-fat” precipitates on corneal endothelium
Iris nodules
Koeppe: clusters of cells on the pupillary border
Busacca: cells on the anterior iris surface
Berlin: cells in the iris angle structure
Miotic pupil
Hypopyon
Hyperemia
Plasmoid iridocyclitis: fibrin with sluggish or no movement of cells in the anterior chamber
Spillover: cells in the anterior vitreous
Low intraocular pressure (IOP) secondary to cyclitis
Elevated IOP secondary to obstruction of the trabecular meshwork, trabeculitis, iris bombè causing pupillary block and angle closure
Cystoid macular edema (CME)
Posterior synechiae
Endothelial dysfunction with associated corneal edema
Fibrin pupillary membrane
edema, fine KPs, and a mid dilated pupil. An association with HLA-B54 has been made with this entity.
History of ocular trauma or surgery
Recent viral or bacterial disease
History of underlying immunologic disease
spondylitis is an aggressive inflammatory arthropathy with a predilection for the central skeleton affecting the sacroiliac joint most severely. The joints between the spine and pelvis, and joints between the vertebrae of the spine, may eventually fuse.
the disease manifests less severely in the female gender. The ratio is thought to be 2 to 3:1 (male:female).
Lower back pain in the morning and after inactivity
Stooped posture
Sleeping in a fetal position to minimize pain
Awakening during the second half of the night because of back pain
Recurrent unilateral uveitis
CBC—may reveal mild anemia
HLA-B27—positive
ESR—may be elevated during active stage
C-reactive protein (CRP)—may be elevated during active stage
Rheumatoid factor—negative
Antinuclear antibodies (ANAs)—negative
Spine and/or pelvis x-ray—look for Romanus lesion—early radiographic sign indicative of disc margin erosion
Technetium bone scan—more sensitive than plain film x-rays
Vitamin D levels—identify patients at risk for osteoporosis
Spine mobility measurements
Electrocardiogram
or cross-reactivity between the microbe and the oral mucosal antigens. In countries with a high prevalence of Behçet’s disease, HLA-B51 has been found in 72% of patients and appears to predispose patients (especially males) to a more severe disease. The association with HLA-B51 has not been demonstrated in the United States. Other trigger factors may include environmental toxins such as heavy metals or pesticides, English walnuts, Gingko nuts, chocolate, and tomatoes.
Recurrent oral ulcerations
Genital sores
Recurrent uveitis
Hypopyon
CBC with differential—anemia observed in some patients with chronic disease
ESR—may be elevated during active stage
CRP—may be elevated during active stage
Cerebral spinal fluid analysis—protein level elevated
Rheumatoid factor—negative
ANAs—negative
IgA—elevated
Complement 3 and 4 levels—elevated
Lipid levels—may be elevated, predisposing the patient to thromboses
Pathergy test (“skin prick”)—following an intradermal puncture to the forearm, a positive test results if the puncture site results in an inflamed and pustular area greater than 2 mm within 24 to 48 h; test is positive in up to 79% of patients with Behçet’s disease although positivity is quite low (˜5%) in white patients limiting the test’s use in many regions
TABLE 10-1 Two Criteria for the Diagnosis of Behçet’s Disease* | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
cataract surgery and vitrectomy. Skin manifestations are treated with topical corticosteroids or antibiotic solutions. The systemic disease is successfully managed with immunosuppressants such as levamisole, colchicine, dapsone, tacrolimus, azathioprine, chlorambucil, cyclosporine A, and cyclophosphamide. Interferon a 2A and B are useful and are thought to have antiviral, immunomodulatory, and antiproliferative properties. TNF-a antagonists, such as infliximab, etanercept, and adalimumab, may be considered for refractory disease including uveitis. Acyclovir may be effective if the etiology is herpes simplex. Exercise, such as swimming or walking, is beneficial to keep joints strong and flexible.
thus presenting with perennial allergic conjunctivitis (PAC).
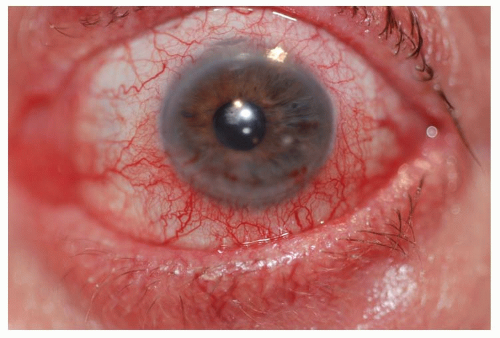 Figure 10-1. Allergic conjunctivitis. (Photo courtesy of Leonid Skorin Jr.) |
Exposure to allergens
Complaint of itching
Personal or family history (first-degree relatives) of atopic disease, that is, allergic rhinitis, bronchial asthma, and atopic dermatitis (AD)
For most patients, the diagnosis of SAC and PAC is clinical and ancillary testing may not be required.
In severe cases of allergic conjunctivitis, superficial conjunctival scrapings may be obtained to evaluate for the presence of eosinophils.
Markers of allergic activity in tear samples may be obtained to assess for levels of IgE, histamine, and tryptase.
Conservative treatment includes the application of cold compresses, artificial tears, and topical ocular vasoconstrictors (Table 10-2). Topical vasoconstrictor/antihistamine combinations cause vascular constriction, decrease vascular permeability, and reduce ocular itching; however, these preparations have
a short duration of action (Table 10-2). Topical ocular antihistamines reduce itching and vasodilation (Table 10-2).
Topical mast cell stabilizers are a safe and effective treatment for chronic allergy symptoms (Table 10-2). A therapeutic effect with these preparations may not be noted for 7 to 14 days; therefore, topical antihistamine/mast cell stabilizers may be indicated to provide rapid symptomatic relief (Table 10-2).
Topical ocular corticosteroids should be reserved for severe cases. However, use judiciously due to potential adverse effects, such as increasing IOP (Table 10-2). Loteprednol is a site-specific steroid with less potential to increase IOP.
Oral antihistamines may provide symptomatic relief from rhinocon-junctivitis. Of note, these drugs may exacerbate dry eye symptoms in some patients (Table 10-2).
Nasal steroid sprays may be a better option than oral antihistamines because they have less systemic effect (Table 10-2).
A topical ocular nonsteroidal anti-inflammatory drug (NSAID) is an alternative treatment option. Ketorolac tromethamine 0.5% is the only NSAID approved for the treatment of SAC (Table 10-2) but is less effective than the other classes of drugs.
TABLE 10-2 Commonly Used Medications to Treat Allergic Conjunctivitis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Prominent signs
Infantile phase (birth to 2 years): these patients present with facial erythema and crusting; extensor extremity lichenification (leathery induration and thickening); exudative papules on the forehead.
Childhood phase (2 to 12 years): patients in this age group present with xerosis (dryness) of skin and lichenification of the flexural extremities.
Adolescent or adult phase (after 12 years): these patients present with chronic relapsing dermatitis, immediate skin test reactivity and oftentimes dermographism.
Ocular signs
Eyelid lichenification
Weeping eczematous lesions
Eversion or stenosis of lacrimal puncta
Dennie-Morgan fold—double lower lid fold
Bilateral keratoconjunctivitis
Papillary conjunctival hypertrophy
Superior corneal shield ulcer
Corneal neovascularization
Symblepharon
Entropion
Trichiasis
Keratoconus
Anterior and posterior subcapsular cataracts in up to 25% of patients
Blepharitis
Atopic keratoconjunctivitis (AKC)
Scarring of the palpebral conjunctiva
Limbal deposits of eosinophils (Trantas’ dots)
Atopic cataracts
Dermatological Evaluation of eczematous lesions to assess the extent of dermatitis and lichenification on the face, scalp, antecubital and popliteal areas, eyelids, neck, outer canthi, and behind the ear lobes. Consultation with a dermatologist is advisable.
Chronic, pruritic, erythematous inflammation of the skin
Asthma
Allergic rhinitis (hay fever)
Serum IgE level and evaluation of skin test reactivity may aid in confirming the diagnosis.
Culture of the eyes with conjunctivitis associated with AD usually grow out Staphylococcus aureus.
Moisturizers, cold compresses, and preventive measures: Treatment should be directed at decreasing xerosis and pruritis with frequent moisturization of the skin with creams (i.e., Eucerin, Cetaphil) or ointments (i.e., white petrolatum jelly or Vaseline). Additionally, cold compresses applied directly to the skin, as needed, decrease itching. Avoidance of sudden changes in temperature or humidity help to decrease symptoms, as does avoidance of sweating or overheating. Scratchy materials (i.e., wool or other irritants) and harsh soaps, detergents, and solvents should also be avoided. Environmental factors that trigger allergies (i.e., pollens, molds, dust mites, and animal dander) should be identified and avoided. For ocular symptoms, artificial tears, instilled four to six times per day, or cold compresses applied to the eyelids may provide relief.
Corticosteroids have been shown to be effective for both skin and ocular symptoms. Relief can be provided by topical corticosteroids during periods of exacerbation when applied sparingly to the affected area, including the periorbital region (Table 10-2). One of the adverse effects of topical corticosteroids is atrophy or thinning of the skin, so use judiciously around the eyes. For severe cases, the addition of topical ocular corticosteroids may be warranted (Table 10-2). Oral corticosteroids are not recommended for treatment of AD, except in very severe cases. Consultation with a dermatologist is recommended.
The topical immunosuppressive/calcineurin inhibitors, pimecrolimus and tacrolimus, have shown excellent results for refractory eczema (Table 10-2). A black box warning, however, has been issued for this class of drugs due to reports of an increased risk of skin malignancy and lymphoma. Consultation with a dermatologist is recommended.
Oral immunosuppressives are reserved for treatment of patients with severe disease in whom conventional therapy is ineffective (Table 10-2). Consultation with a dermatologist is strongly recommended.
Acute ocular symptoms may respond well to topical ocular antihistamines (Table 10-2) or topical ocular antihistamine/mast cell stabilizers (Table 10-2). In addition, topical ocular NSAID can be used as an alternative treatment (Table 10-2).
Chronic ocular symptoms may be managed with topical ocular mast cell stabilizers (Table 10-2) or with the antihistamine/mast cell stabilizer preparations (Table 10-2).
Oral antihistamines: There is little evidence that oral antihistamines are effective in the treatment of AD. Clinically, it may be difficult to distinguish the antipruritic effect of oral antihistamines from the sedative effect since reported improvements in disease severity and quality of life may be due primarily to promotion of restful sleep rather than a reduction in symptoms.
Phototherapy is the supervised use of ultraviolet (UV) light A, B, or a combination of both, by a dermatologist. UV is usually used for mild to moderate cases of AD in adults. It is used only for severe symptoms in children.
Some patients develop a ropy mucous discharge. A papillary conjunctival reaction is common on the tarsal conjunctiva. The lower eyelids are typically more involved than the upper eyelids. Horner-Trantas’ dots may appear as multiple, small, discrete, white spots that form under the epithelial surface at the limbus. These represent accumulations of eosinophils. Chronic tissue changes associated with AKC include conjunctival scarring, fornix foreshortening, and symblepharon formation. Up to 75% of patients develop corneal complications associated with AKC, and these are commonly associated with loss of vision.
Medical history of atopic disease (dermatitis, asthma, rhinitis)
Medical history of multiple allergies including those to food
Ocular history of symptoms of itching, tearing, ropy discharge, burning, photophobia, and decreased vision
Symptomatic relief may be obtained with cold compresses, applied to the eyelids for 10 minutes, four to six times daily, and artificial tears instilled four to six times daily. Maintenance of a cool, moist environment is helpful. In the case of a patient with blepharitis or meibomianitis, the application of warm compresses to the eyelids for 10 minutes, four times daily, and eyelid scrubs, twice daily, provide reduction of symptoms.
Acute ocular symptoms may be controlled with topical ocular antihistamines (Table 10-2), antihistamine/mast cell stabilizers (Table 10-2), or an ocular NSAID (Table 10-2). For patients with AKC, oral antihistamines (Table 10-2) may be needed to control intense itching.
Chronic ocular symptoms can be managed with topical ocular mast cell stabilizers (Table 10-2) or combination of antihistamine/mast cell stabilizers (Table 10-2).
Eyelid eczema may be treated judiciously with a topical corticosteroid ointment or cream (Table 10-2). Topical ocular corticosteroids should also be used judiciously for acute and severe cases (Table 10-2).
Oral corticosteroids are not recommended for the treatment of eyelid eczema, except in very severe cases (Table 10-2). Referral to an allergist or dermatologist is recommended.
Blepharitis or meibomianitis may be treated with topical ocular antibiotics (Table 10-2) or with a topical ocular corticosteroid/antibiotic preparations (Table 10-2). An oral antibiotic, such as doxycycline, may be of clinical utility for chronic posterior blepharitis (Table 10-2).
Excessive mucous production may be treated with a mucolytic agent, such as acetylcysteine (Table 10-2), compounded to a preparation of 10%, formulated by a pharmacist, for topical ocular use.
Topical ocular cyclosporine has been shown to be effective in cases that are refractory to other treatments (Table 10-2). For most severe cases, oral cyclosporine may be used (Table 10-2). However, consultation with an allergist or dermatologist is warranted.
visual axis. Patients may also report foreign body sensation from large papillae or mucus in the tear film. Symptoms typically wax and wane with periods of exacerbation occurring during warmer spring and summer months and decreased symptoms during fall and winter months.
Personal or family history of atopy (allergic rhinitis, AD, or asthma)
Medical history of intense itching, lacrimation, foreign body sensation, blepharospasm, and photophobia
Supportive ocular treatment: As with AKC, VKC may be treated with the application of cold compresses to the eyelids for 10 minutes, four to six times daily, as well as artificial tears four to six times daily. Maintenance of a cool, moist environment is helpful.
Topical ocular corticosteroids: This type of medication is the drug of choice to treat VKC. Up to 85% of patients require its use (Table 10-2).
Topical ocular mast cell stabilizers: These medications have an important role in managing chronic cases of VKC (Table 10-2). Clinical improvement may be delayed with mast cell
stabilizers; therefore, a topical ocular antihistamine (Table 10-2), antihistamine/mast cell stabilizer (Table 10-2), or corticosteroid (Table 10-2) can be used concomitantly to relieve symptoms. Intractable cases of VKC may respond to a combination of oral aspirin (Table 10-2) and topical ocular cromolyn sodium 2% twice daily.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


