10 IMMUNE DISEASE
Anterior Uveitis
Richard F. Multack, Samuel J. Multack and Leonid Skorin Jr
ICD-9: 364.0
 THE DISEASE
THE DISEASE
Pathophysiology
The uvea consists of the middle, pigmented, vascular structures of the eye and includes the iris, ciliary body, and choroid. Uveitis refers to inflammation of the uveal tract. Anterior uveitis can be divided into iritis, anterior cyclitis (ciliary body inflammation), and iridocyclitis. Anterior uveitis does not include involvement of the pars plana or the posterior segment. Adjacent, nonvascular structures such as the cornea (keratouveitis) and sclera (sclerouveitis) are often affected secondarily in the inflammatory process.
Anterior uveitis can also be divided by its clinical course. Acute uveitis refers to inflammation that lasts for weeks or a few months and resolves once the attack is over. Chronic uveitis may last for many months or years without clearing completely between exacerbations. Uveitis can be classified by its etiology, infectious or noninfectious. Most commonly anterior uveitis is a noninfectious/sterile process.
Acute and chronic uveitis can be further subdivided into granulomatous and nongranulomatous that is determined by clinical findings. Granulomatous uveitis presents with large, greasy, “mutton-fat” precipitates on the corneal endothelium with large clumps of inflammatory cells present in the anterior chamber because of exuberant macrophage activity. Nongranulomatous uveitis presents with fine corneal endothelial precipitates and anterior chamber activity.
Etiology
The causes of anterior uveitis are multiple and varied and commonly go undetected. Most types of anterior uveitis are sterile inflammatory reactions. Underlying associations include ankylosing spondylitis, reactive arthritis (Reiter’s syndrome), Adamantiades-Behçet’s disease (ABD), trauma, psoriatic arthritis, juvenile rheumatoid arthritis/juvenile idiopathic arthritis (JRA/JIA), postoperative iritis, glaucomatocyclitic crisis, intraocular lens–induced UGH syndrome (uveitis-glaucoma-hyphema), Fuchs heterochromic iridocyclitis (FHI), sarcoidosis, and idiopathic causes. Infectious causes of anterior uveitis may include Lyme disease, various viral etiologies, syphilis, and tuberculosis.
The Patient
The signs and symptoms can vary and depend on the anatomical involvement, onset, and duration of the uveitis and whether it is granulomatous or nongranulomatous.
Clinical Symptoms
The patient with an acute anterior uveitis will complain of ocular pain, red eye, photophobia, epiphora, and blurred vision. Pain from anterior uveitis will often be referred to the eyebrow area. These symptoms may be very mild or absent in chronic anterior uveitis. Characteristic HLA-B27-associated uveitis is recurrent with alternating bilateral involvement.
Clinical Signs
- Cells and flare in the anterior chamber
- Circumcorneal ciliary flush
- Nongranulomatous: fine keratic precipitates (KPs) on corneal endothelium
- Granulomatous: “mutton-fat” precipitates on corneal endothelium
- Iris nodules
Koeppe: clusters of cells on the pupillary border
Busacca: cells on the anterior iris surface
Berlin: cells in the iris angle structure
- Miotic pupil
- Hypopyon
- Hyperemia
- Plasmoid iridocyclitis: fibrin with sluggish or no movement of cells in the anterior chamber
- Spillover: cells in the anterior vitreous
- Low intraocular pressure (IOP) secondary to cyclitis
- Elevated IOP secondary to obstruction of the trabecular meshwork, trabeculitis, iris bombè causing pupillary block and angle closure
- Cystoid macular edema (CME)
- Posterior synechiae
- Endothelial dysfunction with associated corneal edema
- Fibrin pupillary membrane
Demographics
Anterior uveitis is the most common form of uveitis with an incidence of 9/100,000 people and increasing to 100/100,000 in patients over 65 years of age. Most patients affected are from 20 to 50 years of age. Some forms of anterior uveitis, such as acute nongranulomatous uveitis secondary to ankylosing spondylitis and reactive arthritis (Reiter’s syndrome), are more often found in men. Chronic anterior uveitis is more common in women. Immunologic and hormonal differences are thought to underlie some specific types of uveitis.
Seronegative spondyloarthropathies (rheumatoid factor [RF] negative) that are associated with acute anterior uveitis are often HLA-B27 positive (located on the short arm of chromosome 6). HLA-B27 is positive in 1.4% to 8% of the general population. In patients who present with acute iritis, up to 60% prove to be HLA-B27 positive. Ankylosing spondylitis, reactive arthritis, inflammatory bowel disease (IBD), and psoriatic arthritis are all included in the seronegative spondyloarthropathies.
ABD, is associated with HLA-B51. It is a perivascular inflammation of unknown cause that results in a generalized occlusive vasculitis. The Behçet’s triad includes a hypopyon with iritis, aphthous stomatitis, and genital ulceration. It is less common in the United States and is more common in Japan and Eastern Europe. Researchers in Japan have suggested a diagnostic system that includes major and minor criteria. Major criteria are recurrent oral ulcers, skin lesions, recurrent genital ulcers, and ocular inflammatory disease. Minor criteria include epididymitis, arthritis, vasculitis, gastrointestinal ulcerations, and psychiatric symptoms. Based on the number of criteria, different types have been developed including complete, incomplete, suspect, and possible. Eye findings may include a nongranulomatous anterior uveitis, retinal vasculitis, retinal hemorrhage, macular edema, retinal necrosis, ischemic optic neuropathy, and vitritis. Systemic signs may include erythema nodosum, nondestructive arthritis, IBD, mucous membrane ulcerations, pericarditis, myocarditis, and central nervous system (CNS) strokes.
Glaucomatocyclitic crisis (Posner-Schlossman’s syndrome) is a mild to moderate uveitis associated with mid to upper range elevated IOP, corneal edema, fine KPs, and a mid dilated pupil. An association with HLA-B54 has been made with this entity.
Lens-associated uveitis or phacoantigenic endophthalmitis is a presumed immune response to lens protein after violation of the lens capsule.
Postoperative uveitis can include infection, lens-associated and intraocular lens (IOL)-associated inflammation. Infectious causes can include Staphylococcus epidermidis, Candida, and delayed endophthalmitis caused by Propionibacterium acnes. Manipulation of the anterior chamber results in some break down of the blood–aqueous barrier (BAB) leading to increased risk of uveitis.
Pseudophakic uveitis may be seen with closed loop anterior chamber intraocular lenses although it is much less common with newer flexible intraocular lens implants.
FHI or Fuchs uveitis syndrome is usually unilateral with varying symptoms of mild discomfort to none. The diagnosis is based on heterochromia, lack of synechiae, and diffusely distributed KPs.
Masquerade syndromes, such as syphilis, may present with an anterior uveitis. Syphilis accounts for 1% to 3% of all uveitis cases. Patients will develop anterior uveitis in 5% to 10% of secondary syphilis cases.
Significant History
- History of ocular trauma or surgery
- Recent viral or bacterial disease
- History of underlying immunologic disease
Ancillary Tests
Ocular assessment includes visual acuity, pupil testing, IOP, slit-lamp examination, and dilated fundus evaluation.
Laboratory studies: complete blood count (CBC) with differential, erythrocyte sedimentation rate (ESR), chest and pelvis x-ray, Lyme titer, antinuclear-antibody, rapid plasma reagin (RPR) or VRDL, FTA-ABS, purified protein derivative (PPD), human leukocyte antigen (HLA) typing, angiotension converting enzyme, and serum lysozyme.
The Treatment
As the etiology of anterior uveitis is often unknown, treatment is directed to control the inflammation and to prevent damage to uveal vasculature that can result in chronic recalcitrant uveitis or secondary side effects, such as posterior synechiae, cataracts, and glaucoma.
Cycloplegic agents are used to relieve pain, decrease synechiae formation, and reduce the permeability of the iris vasculature. Cyclopentolate or homatropine can be used for mild to moderate inflammation, whereas scopolamine or atropine may be used for more severe inflammation.
Corticosteroids can be applied topically, by periocular injection, systemically, or by intravitreal implants such as fluocinolone acetonide/Retisert (Bausch and Lomb). The steroid therapy should never be stopped abruptly because this may lead to severe rebound in inflammation.
Patients who fail or are intolerant to steroids may require immunomodulatory agents, such as methotrexate or cyclosporine and azathioprine. Recently, tumor necrosis factor-alpha (TNF-α) inhibitors such as infliximab have shown promising results.
Ankylosing Spondylitis
Tammy P. Than
ICD-9: 720.0M
 THE DISEASE
THE DISEASE
Pathophysiology
Ankylosing spondylitis belongs to a group of seronegative spondyloarthropathies in which the RF is absent. Ankylosing spondylitis is an aggressive inflammatory arthropathy with a predilection for the central skeleton affecting the sacroiliac joint most severely. The joints between the spine and pelvis, and joints between the vertebrae of the spine, may eventually fuse.
Etiology
The etiology is unknown but is thought to be multifactorial. HLA-B27 has been found in 93% of patients with ankylosing spondylitis while present in only 6% of controls. In the absence of HLA-B27, genes for other inflammatory conditions may be important in predisposing individuals. Evidence for this is the greater association with other inflammatory conditions such as Crohn’s disease or psoriasis. Alternatively, bacterial and viral infections may trigger ankylosing spondylitis. In particular, Klebsiella pneumoniae, commonly found in the gastrointestinal tract, is more common in patients with the disease compared to controls. There appears to be a familial pattern as there is a higher incidence of ankylosing spondylitis if there is a positive history in a first-degree relative.
The Patient
Clinical Symptoms
Symptoms usually begin in the early 20s with an onset rarely after the age of 40. Systemic symptoms begin with intermittent hip and/or lower back pain that are worse at night or after inactivity. Bent posture eases the pain, which improves later in the day and with exercise. There may be pain or tenderness in the ribs, shoulder blades, hips, thighs, shins, and heels. The patient may also complain of fatigue, loss of appetite, and general discomfort. Ocular symptoms include pain, redness, and photophobia if uveitis is present.
The disease in women tends to be less severe than in men and may present with neck pain and breast pain in the absence of lower back pain.
Clinical Signs
Uveitis occurs in about 25% of patients with ankylosing spondylitis presenting as an acute, unilateral, nongranulomatous episode. The ocular manifestations may occur prior to joint and skeletal signs, and ankylosing spondylitis accounts for 10% to 33% of all cases of anterior uveitis.
Early on, limited flexibility of the lower spine is noted. Enthesopathy, new bone formation at the attachment of tendons and ligaments, may be noted. Over time, ankylosing spondylitis progresses to involve the entire spine—lordosis (forward curvature) of the lumbar spine, kyphosis (excessive curvature with convexity backward) of the thoracic spine, and hyperextension of the cervical spine. Other joints are involved in about 33% of cases, most frequently presenting with inflammation of the hips, knees, ankles, and shoulders. Osteoporosis is noted in early and late stages of the disease in 19% to 62% of cases and may result in vertebral fractures. Tendons and ligaments can also become inflamed. Cardiac complications include valvular disease (e.g., aortic valve stenosis) and aortitis. Ankylosing spondylitis can affect the bones of the rib cage, reducing lung capacity. Patients may have signs of psoriasis or IBD.
Demographics
The incidence of ankylosing spondylitis is 0.21% in Americans over the age of 15. The incidence is greatest among Native American Indians and Eskimos, and it is rare in African Americans. The peak age of onset is in the mid-20s, although a juvenile form develops around age 8 to 10 years. The male to female ratio is often overestimated because women are frequently underdiagnosed because the disease manifests less severely in the female gender. The ratio is thought to be 2 to 3:1 (male:female).
Significant History
- Lower back pain in the morning and after inactivity
- Stooped posture
- Sleeping in a fetal position to minimize pain
- Awakening during the second half of the night because of back pain
- Recurrent unilateral uveitis
Ancillary Tests
- CBC—may reveal mild anemia
- HLA-B27—positive
- ESR—may be elevated during active stage
- C-reactive protein (CRP)—may be elevated during active stage
- Rheumatoid factor—negative
- Antinuclear antibodies (ANAs)—negative
- Spine and/or pelvis x-ray—look for Romanus lesion—early radiographic sign indicative of disc margin erosion
- Technetium bone scan—more sensitive than plain film x-rays
- Vitamin D levels—identify patients at risk for osteoporosis
- Spine mobility measurements
- Electrocardiogram
The Treatment
Exercises to strengthen the back muscles to maintain erect posture, to maintain chest expansion, and to maintain or improve spinal mobility should be part of the patient’s daily routine. Physical therapy is also useful. Heat applied to the joints helps reduce joint pain unless active inflammation is present in which case cold packs are preferred. The patient should be encouraged to sleep on a firm mattress with good neck support. A smoking cessation program, if pertinent, should be a priority. Oral nonsteroidal anti-inflammatory agents are the first line of therapeutic intervention. More aggressive management includes sulfasalazine and other disease-modifying antirheumatic drugs. TNF-α antagonists, such as etanercept, infliximab, and adalimumab, are useful if conventional treatment fails. Intra-articular corticosteroid injections may be beneficial. Topical corticosteroids and cycloplegics are used to manage the anterior uveitis. Approximately 90% of patients remain fully independent with little or no disability.
Behçet’s Disease
Tammy P. Than
ICD-9: 136.1
 THE DISEASE
THE DISEASE
Pathophysiology
Behçet’s disease is a rare, nongranulomatous, obliterative vasculitis that is thought to be immune mediated. Antiendothelial cell antibodies are detected in an increased prevalence and neutrophils are hyperactive interacting with T cells. Activated T cells produce TNF-α which results in increased levels of proinflammatory cytokines. It affects arteries and veins resulting in thrombosis. The disease is chronic and multisystemic, characterized by oral and genital mucocutaneous ulcerations, skin rashes, arthritis, thrombophlebitis, uveitis, colitis, and neurologic symptoms.
Etiology
The exact etiology is unknown; however, immune regulation, immunogenetics, vascular abnormalities, or bacterial or viral infection may play a role in the development of Behçet’s disease. An infectious cause may be associated with herpes simplex virus I or Streptococcus sanguis in which there is autoimmunity or cross-reactivity between the microbe and the oral mucosal antigens. In countries with a high prevalence of Behçet’s disease, HLA-B51 has been found in 72% of patients and appears to predispose patients (especially males) to a more severe disease. The association with HLA-B51 has not been demonstrated in the United States. Other trigger factors may include environmental toxins such as heavy metals or pesticides, English walnuts, Gingko nuts, chocolate, and tomatoes.
The Patient
Clinical Symptoms
Prior to the onset of Behçet’s disease, the patient may experience a prodrome and report malaise, weight loss, anorexia, general weakness, and headache. During the disease process, the patient will complain of a painful mouth and genital sores, photophobia with a red and painful eye, and joint pain.
Clinical Signs
Behçet’s disease waxes and wanes with periods of exacerbation and remission. As time passes, the recurrence frequency decreases and the severity diminishes.
Ocular signs are present in 75% of patients. Bilateral nongranulomatous uveitis, which may include a small hypopyon, usually follows the onset of oral ulcers by 3 to 4 years, but ocular disease is the initial presentation in 20% of patients. Late-stage ocular complications include neovascularization, glaucoma, cataracts, retinal detachment, vascular occlusive disease, and optic atrophy. Other potential ocular manifestations are sixth-nerve palsy, hemiparesis, visual field loss, idiopathic intracranial hypertension, internuclear ophthalmoplegia, and complications from intracranial artery aneurysms.
There are two common criteria for the diagnosis of Behçet’s disease. The International Study Group (ISG; 1990) criteria require the presence of oral ulceration, which may be a limitation. Therefore, some recommend applying the 1987 criteria from the Japanese group in conjunction with the ISG guidelines (Table 10-1).
TABLE 10-1 Two Criteria for the Diagnosis of Behçet’s Disease*
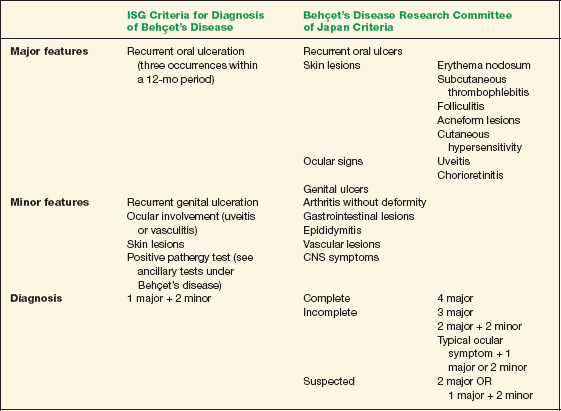
Data from Behçet’s Disease Research Committee of Japan. Behçet’s disease: Guide to diagnosis of Behçet’s disease. Jpn J Ophthalmol 1974:18:291–294; and International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet 1990;335:1078–1080.
Demographics
Behçet’s disease has a worldwide distribution and is most prevalent (and more virulent) in the Mediterranean, Middle East, and Far East with an estimated incidence of 14 to 380/100,000. The prevalence in the United States is 0.33/100,000. Those from high-risk areas who immigrate to low prevalence areas have an intermediate risk, which suggests that environmental factors play a role in the disease. The predominant age at diagnosis is in the third to fourth decades, with men affected more often and with a more severe course in some regions. Prognosis is poorer with neurological involvement, where there is a 20% mortality rate after 7 years. Chronic morbidity is usual, with ophthalmic involvement as the leading cause. Blindness occurs in up to 25% of eyes within 10 years of onset of ocular disease. The effects of the disease appear to be cumulative.
Significant History
- Recurrent oral ulcerations
- Genital sores
- Recurrent uveitis
- Hypopyon
Ancillary Tests
Ocular evaluation includes assessment of the anterior chamber and IOP. A dilated fundus examination should be performed to rule out occlusive disease and its sequelae, vasculitis, retinal detachment, and optic nerve disease.
There is no diagnostic laboratory test specific for Behçet’s disease, but the following may be helpful:
- CBC with differential—anemia observed in some patients with chronic disease
- ESR—may be elevated during active stage
- CRP—may be elevated during active stage
- Cerebral spinal fluid analysis—protein level elevated
- Rheumatoid factor—negative
- ANAs—negative
- IgA—elevated
- Complement 3 and 4 levels—elevated
- Lipid levels—may be elevated, predisposing the patient to thromboses
- Pathergy test (“skin prick”)—following an intradermal puncture to the forearm, a positive test results if the puncture site results in an inflamed and pustular area greater than 2 mm within 24 to 48 h; test is positive in up to 79% of patients with Behçet’s disease although positivity is quite low (~5%) in white patients limiting the test’s use in many regions
- CRP—may be elevated during active stage
The Treatment
There is no established standard therapeutic regimen for Behçet’s disease, but treatment is guided by organ involvement. Prognosis has improved in the past decade with more aggressive treatment strategies.
Uveitis is managed with topical corticosteroids and cycloplegia. Laser photocoagulation may be used to manage retinal neovascularization. Ocular management may also include cataract surgery and vitrectomy. Skin manifestations are treated with topical corticosteroids or antibiotic solutions. The systemic disease is successfully managed with immunosuppressants such as levamisole, colchicine, dapsone, tacrolimus, azathioprine, chlorambucil, cyclosporine A, and cyclophosphamide. Interferon α 2A and B are useful and are thought to have antiviral, immunomodulatory, and antiproliferative properties. TNF-α antagonists, such as infliximab, etanercept, and adalimumab, may be considered for refractory disease including uveitis. Acyclovir may be effective if the etiology is herpes simplex. Exercise, such as swimming or walking, is beneficial to keep joints strong and flexible.
Allergic Conjunctivitis
Salisa K. Williams and Daniel Ullmann
The ocular surface may present an immunologic response as an inflammation of the conjunctiva and cornea. According to the Gell and Coombs classification system for immunologic reactions, four types of hypersensitivity reactions are recognized:
Type I: Immunoglobulin E (IgE)-mediated. This is also known as immediate hypersensitivity that can manifest as an anaphylactic (systemic) or atopic (local) reaction. The antigen binds to the antigen-binding fragment (Fab) portion of the IgE on either mast cells or basophils. Mast cells produce the initial reaction, while basophils initiate the late phase reaction. The cross-linking causes degranulation and the release of histamine resulting in increased vascular permeability and smooth muscle contraction.
Type II: Antibody-mediated (cytotoxic). Circulating antibodies react with antigens on the surface of cells, which leads to cell damage by either phagocytosis or complement activation. These cytotoxic reactions appear to be the underlying etiology of cicatricial pemphigoid (CP) and Mooren ulcer.
Type III: Immune-complex mediated. Soluble antigen-antibody complexes form when the antigen is abundant. Complex deposition in tissues causes type III hypersensitivity. The Arthus reaction is a classic systemic type III reaction, while Wesley rings are an example of a corneal type III reaction.
Type IV: Cell-mediated (delayed type). Presentation of antigen to CD4+ T cells leads to tissue damage via the release of cytokines and the subsequent accumulation of neutrophils and macrophages. Ocular examples of type IV hypersensitivity include drug allergies, corneal allograft rejection, and contact dermatitis.
This section presents the major hypersensitivity conditions involving the conjunctiva, more commonly referred as allergic conjunctivitis.
Seasonal Allergic Conjunctivitis and Perennial Allergic Conjunctivitis
ICD-9: 477.9
 THE DISEASE
THE DISEASE
The conjunctiva is a thin mucous membrane that lines the posterior surface of the eyelids and extends over the globe to the corneal limbus. It has a rich vascular supply and is an important, immunologically active defense against pathogens. Seasonal allergic conjunctivitis (SAC) is a mild to moderate manifestation of the immune response of the conjunctiva to a wide variety of antigens. Patients may experience symptoms on a seasonal basis when exposed to antigens or may exhibit symptoms year round, thus presenting with perennial allergic conjunctivitis (PAC).
Pathophysiology
Once primary exposure and sensitization to antigens occur, repeat exposure to the antigen initiates the inflammatory response. Mast cells migrate into the conjunctival epithelium and are the primary cell type involved in the inflammatory response. Degranulation of mast cells and basophils releases histamine, bradykinins, and leukotrienes. These primary inflammatory mediators induce vasodilatation, tissue edema, and nerve stimulation.
Etiology
Among patients with allergic conjunctivitis, airborne allergens such as ragweed, pollens, dander, dust, or mold spores initiate the immune response resulting in symptoms of allergic conjunctivitis. Patients with SAC present with symptoms in the spring, summer, and fall due to the abundance of airborne allergens found in tree pollen, grass pollen, and weed pollen, respectively.
Patients with PAC may present symptoms throughout the year; thus, in addition to the seasonal allergens, other allergens such as dust mites, cockroaches, and pet dander play a role in the development of symptoms among these patients.
The Patient
Clinical Symptoms
Patients with SAC and PAC complain of conjunctival hyperemia, burning, tearing, and ocular itching. Symptoms are transient, recurrent, and often follow seasonal patterns. The timing of the symptoms differentiates between patients with SAC and patients with PAC.
Clinical Signs
Patients develop variable degrees of conjunctival hyperemia, chemosis (edema), and eyelid edema (Fig. 10-1). In some patients, a papillary conjunctival response may be observed. The conjunctiva often appears milky due to the edema.
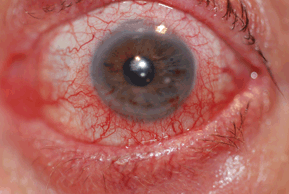
Figure 10-1. Allergic conjunctivitis.
(Photo courtesy of Leonid Skorin Jr.)
Demographics
SAC and PAC are common conditions affecting all segments of the population.
Significant History
- Exposure to allergens
- Complaint of itching
- Personal or family history (first-degree relatives) of atopic disease, that is, allergic rhinitis, bronchial asthma, and atopic dermatitis (AD)
Ancillary Tests
- For most patients, the diagnosis of SAC and PAC is clinical and ancillary testing may not be required.
- In severe cases of allergic conjunctivitis, superficial conjunctival scrapings may be obtained to evaluate for the presence of eosinophils.
- Markers of allergic activity in tear samples may be obtained to assess for levels of IgE, histamine, and tryptase.
The Treatment
1. Conservative treatment includes the application of cold compresses, artificial tears, and topical ocular vasoconstrictors (Table 10-2). Topical vasoconstrictor/antihistamine combinations cause vascular constriction, decrease vascular permeability, and reduce ocular itching; however, these preparations have a short duration of action (Table 10-2). Topical ocular antihistamines reduce itching and vasodilation (Table 10-2).
2. Topical mast cell stabilizers are a safe and effective treatment for chronic allergy symptoms (Table 10-2). A therapeutic effect with these preparations may not be noted for 7 to 14 days; therefore, topical antihistamine/mast cell stabilizers may be indicated to provide rapid symptomatic relief (Table 10-2).
3. Topical ocular corticosteroids should be reserved for severe cases. However, use judiciously due to potential adverse effects, such as increasing IOP (Table 10-2). Loteprednol is a site-specific steroid with less potential to increase IOP.
4. Oral antihistamines may provide symptomatic relief from rhinoconjunctivitis. Of note, these drugs may exacerbate dry eye symptoms in some patients (Table 10-2).
5. Nasal steroid sprays may be a better option than oral antihistamines because they have less systemic effect (Table 10-2).
6. A topical ocular nonsteroidal anti-inflammatory drug (NSAID) is an alternative treatment option. Ketorolac tromethamine 0.5% is the only NSAID approved for the treatment of SAC (Table 10-2) but is less effective than the other classes of drugs.
TABLE 10-2 Commonly Used Medications to Treat Allergic Conjunctivitis
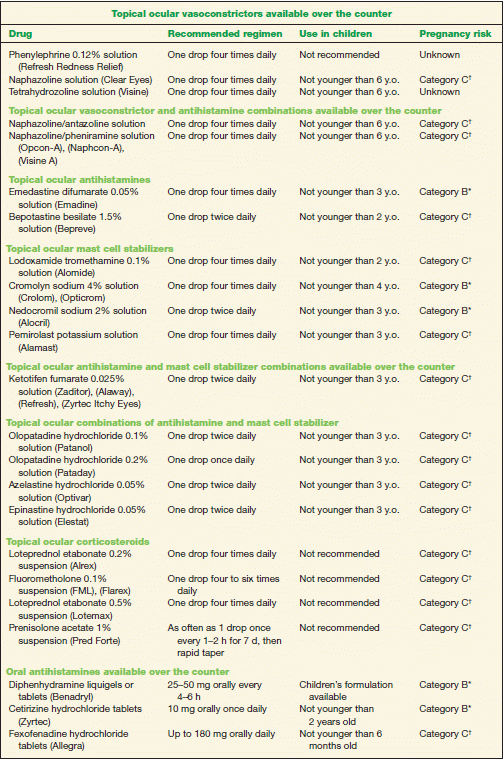
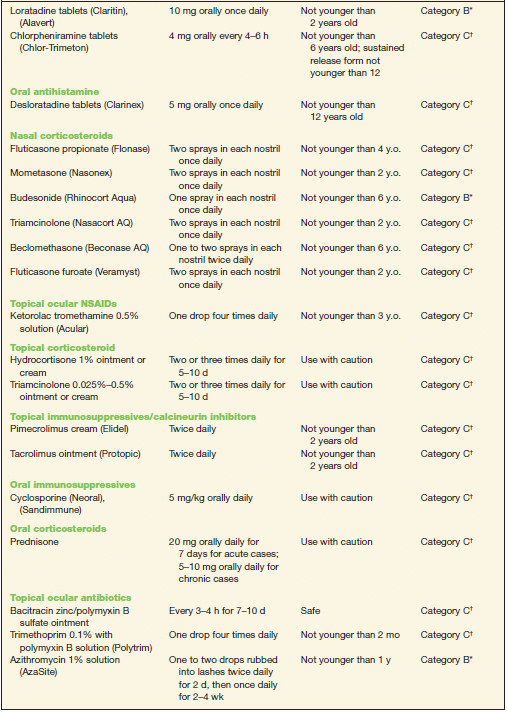
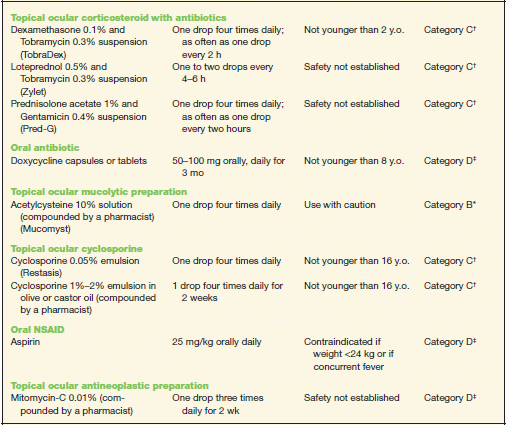
* Category B denotes: presumed safe, animal studies.
† Category C denotes: uncertain safety, animal studies show an adverse effect.
‡ Category D denotes: unsafe, use may be justifiable in certain circumstances.
Atopic Dermatitis
ICD-9: 691.8
 THE DISEASE
THE DISEASE
AD is a chronic, relapsing inflammatory condition that often presents with ocular involvement. This disorder emanates from a complex interaction among various susceptible genes, defects in skin barrier function, immunological responses, host and environmental factors, and infectious agents. The atopic triad consists of AD in association with allergic rhinitis and asthma.
Pathophysiology
Approximately 80% of patients with AD have elevated serum levels of IgE. This increase in the concentration of IgE is more pronounced when other atopic disorders are present. It appears that patients with AD have an overactive type 2 T-helper (Th2) cell cytokine immune response, which stimulates B-cells to produce IgE. In addition, patients with AD have defective cell-mediated immunity as manifested by an increased susceptibility to viral and fungal infections, which are also present in atopy (AD, allergic rhinitis and asthma). Decreased numbers and diminished function of T lymphocytes usually correlate with disease activity. Defective chemotactic activity of neutrophils and monocytes is also found among patients with AD.
Etiology
Atopy (AD, allergic rhinitis, and asthma) is a form of hypersensitivity reaction whose immunologic factors relate to potential genetically mediated defects in metabolism or biochemical response to exogenous substances.
The Patient
Most patients develop AD before the age of 5 years. Symptoms and signs of atopy include pruritic, eczematous lesions, which typically present on the face, scalp, antecubital and popliteal areas, eyelids, neck, outer canthi, and behind the ear lobes.
Clinical Symptoms
The patient with atopy will complain of intense pruritus (itching) of the skin and the eyes with partial relief from rubbing.
Clinical Signs
1. Prominent signs
- Infantile phase (birth to 2 years): these patients present with facial erythema and crusting; extensor extremity lichenification (leathery induration and thickening); exudative papules on the forehead.
- Childhood phase (2 to 12 years): patients in this age group present with xerosis (dryness) of skin and lichenification of the flexural extremities.
- Adolescent or adult phase (after 12 years): these patients present with chronic relapsing dermatitis, immediate skin test reactivity and oftentimes dermographism.
2. Ocular signs
- Eyelid lichenification
- Weeping eczematous lesions
- Eversion or stenosis of lacrimal puncta
- Dennie-Morgan fold—double lower lid fold
- Bilateral keratoconjunctivitis
- Papillary conjunctival hypertrophy
- Superior corneal shield ulcer
- Corneal neovascularization
- Symblepharon
- Entropion
- Trichiasis
- Keratoconus
- Anterior and posterior subcapsular cataracts in up to 25% of patients
- Blepharitis
- Atopic keratoconjunctivitis (AKC)
- Scarring of the palpebral conjunctiva
- Limbal deposits of eosinophils (Trantas’ dots)
- Atopic cataracts
- Atopic keratoconjunctivitis (AKC)
3. Dermatological Evaluation of eczematous lesions to assess the extent of dermatitis and lichenification on the face, scalp, antecubital and popliteal areas, eyelids, neck, outer canthi, and behind the ear lobes. Consultation with a dermatologist is advisable.
Demographics
The prevalence of AD has been estimated from 10% to 20% in children and from 1% to 3% among adults. It typically develops before 5 years of age (90% of cases) and most often by 1 year of age (60%). There is a family history of AD, asthma, or hay fever in 70% of patients. About 40% of patients experience a spontaneous remission by age 5 years.
Significant History
- Chronic, pruritic, erythematous inflammation of the skin
- Asthma
- Allergic rhinitis (hay fever)
Ancillary Tests
- Serum IgE level and evaluation of skin test reactivity may aid in confirming the diagnosis.
- Culture of the eyes with conjunctivitis associated with AD usually grow out Staphylococcus aureus.
The Treatment
1. Moisturizers, cold compresses, and preventive measures: Treatment should be directed at decreasing xerosis and pruritis with frequent moisturization of the skin with creams (i.e., Eucerin, Cetaphil) or ointments (i.e., white petrolatum jelly or Vaseline). Additionally, cold compresses applied directly to the skin, as needed, decrease itching. Avoidance of sudden changes in temperature or humidity help to decrease symptoms, as does avoidance of sweating or overheating. Scratchy materials (i.e., wool or other irritants) and harsh soaps, detergents, and solvents should also be avoided. Environmental factors that trigger allergies (i.e., pollens, molds, dust mites, and animal dander) should be identified and avoided. For ocular symptoms, artificial tears, instilled four to six times per day, or cold compresses applied to the eyelids may provide relief.
2. Corticosteroids have been shown to be effective for both skin and ocular symptoms. Relief can be provided by topical corticosteroids during periods of exacerbation when applied sparingly to the affected area, including the periorbital region (Table 10-2). One of the adverse effects of topical corticosteroids is atrophy or thinning of the skin, so use judiciously around the eyes. For severe cases, the addition of topical ocular corticosteroids may be warranted (Table 10-2). Oral corticosteroids are not recommended for treatment of AD, except in very severe cases. Consultation with a dermatologist is recommended.
3. The topical immunosuppressive/calcineurin inhibitors, pimecrolimus and tacrolimus, have shown excellent results for refractory eczema (Table 10-2). A black box warning, however, has been issued for this class of drugs due to reports of an increased risk of skin malignancy and lymphoma. Consultation with a dermatologist is recommended.
4. Oral immunosuppressives are reserved for treatment of patients with severe disease in whom conventional therapy is ineffective (Table 10-2). Consultation with a dermatologist is strongly recommended.
5. Acute ocular symptoms may respond well to topical ocular antihistamines (Table 10-2) or topical ocular antihistamine/mast cell stabilizers (Table 10-2). In addition, topical ocular NSAID can be used as an alternative treatment (Table 10-2).
6. Chronic ocular symptoms may be managed with topical ocular mast cell stabilizers (Table 10-2) or with the antihistamine/mast cell stabilizer preparations (Table 10-2).
7. Oral antihistamines: There is little evidence that oral antihistamines are effective in the treatment of AD. Clinically, it may be difficult to distinguish the antipruritic effect of oral antihistamines from the sedative effect since reported improvements in disease severity and quality of life may be due primarily to promotion of restful sleep rather than a reduction in symptoms.
8. Phototherapy is the supervised use of ultraviolet (UV) light A, B, or a combination of both, by a dermatologist. UV is usually used for mild to moderate cases of AD in adults. It is used only for severe symptoms in children.
Atopic Keratoconjunctivitis
ICD-9: 372.05
 THE DISEASE
THE DISEASE
AKC is a rare but potentially blinding condition characterized by bilateral and symmetric conjunctivitis more prevalent in men than in women. In addition, patients with AKC may present with affected eyelid skin, lid margins, as well as affected cornea and lens. Among patients with AKC, 95% have a long standing history of AD, while 87% have a history of asthma. Decreased vision and blindness are the results of superficial punctate keratitis, epithelial defects, corneal scarring/thinning, keratoconus, cataracts, and symblepharon formation.
Pathophysiology
AKC is a perennial disorder characterized by both type I and type IV hypersensitivity reactions; thus, the inflammatory response of AKC has features of immediate and delayed changes of the conjunctiva and cornea. The serum and tear IgE levels are elevated during exacerbated periods of AKC. Among these patients, the prevalence of AD is approximately 3%, with 25% of them having ocular disease.
Etiology
The etiology of AKC remains unknown. Approximately, 5% of patients present with childhood vernal keratoconjunctivitis (VKC). In addition, patients with AKC have a family history of allergic disease, hence atopy. Patients with atopy often present with environmental allergies, asthma (allergic), rhinitis, and AD.
The Patient
Clinical Symptoms
Patients present with bilateral, symmetric itching and tearing. While some patients experience seasonal exacerbations, others are able to identify allergens associated with an increase in symptoms. In cases of corneal ulceration or erosion, pain and photophobia may occur. Late symptoms may include reduced vision from corneal scarring.
Clinical Signs
The conjunctiva develops hyperemia and can become noticeably thickened. Some patients develop a ropy mucous discharge. A papillary conjunctival reaction is common on the tarsal conjunctiva. The lower eyelids are typically more involved than the upper eyelids. Horner-Trantas’ dots may appear as multiple, small, discrete, white spots that form under the epithelial surface at the limbus. These represent accumulations of eosinophils. Chronic tissue changes associated with AKC include conjunctival scarring, fornix foreshortening, and symblepharon formation. Up to 75% of patients develop corneal complications associated with AKC, and these are commonly associated with loss of vision.
Corneal findings include persistent keratopathy, corneal neovascularization, corneal scarring, and the development of keratoconus. AKC has also been associated with a higher incidence of cortical cataract formation (10% of cases). Patients with AKC have depressed T-cell function and may develop staphylococcal blepharitis, meibomianitis, and herpetic keratitis as secondary complications. Rhegmatogenous retinal detachment (equivalent to traumatic detachment) has been linked to AKC attributed to vitreal degeneration secondary to excessive eye rubbing practice by patients to alleviate symptoms of itching.
Demographics
Patients with AKC range from late teens to 50 years of age, with peak incidence between the ages of 30 and 50. This condition occurs more often in men than in women. Among patients with AD, the prevalence of AKC is from 25% to 40%.
Significant History
- Medical history of atopic disease (dermatitis, asthma, rhinitis)
- Medical history of multiple allergies including those to food
- Ocular history of symptoms of itching, tearing, ropy discharge, burning, photophobia, and decreased vision
Ancillary Tests
Ancillary tests are generally not useful in diagnosing AKC. Conjunctival biopsy can help differentiate AKC from CP. Conjunctival biopsy results reveal excessive eosinophils, mast cells, and goblet cells. Conjunctival biopsy can also help to histologically differentiate AKC from CP by the presence of basement membrane antibodies or complement components in CP.
The Treatment
1. Symptomatic relief may be obtained with cold compresses, applied to the eyelids for 10 minutes, four to six times daily, and artificial tears instilled four to six times daily. Maintenance of a cool, moist environment is helpful. In the case of a patient with blepharitis or meibomianitis, the application of warm compresses to the eyelids for 10 minutes, four times daily, and eyelid scrubs, twice daily, provide reduction of symptoms.
2. Acute ocular symptoms may be controlled with topical ocular antihistamines (Table 10-2), antihistamine/mast cell stabilizers (Table 10-2), or an ocular NSAID (Table 10-2). For patients with AKC, oral antihistamines (Table 10-2) may be needed to control intense itching.
3. Chronic ocular symptoms can be managed with topical ocular mast cell stabilizers (Table 10-2) or combination of antihistamine/mast cell stabilizers (Table 10-2).
4. Eyelid eczema may be treated judiciously with a topical corticosteroid ointment or cream (Table 10-2). Topical ocular corticosteroids should also be used judiciously for acute and severe cases (Table 10-2). Oral corticosteroids are not recommended for the treatment of eyelid eczema, except in very severe cases (Table 10-2). Referral to an allergist or dermatologist is recommended.
5. Blepharitis or meibomianitis may be treated with topical ocular antibiotics (Table 10-2) or with a topical ocular corticosteroid/antibiotic preparations (Table 10-2). An oral antibiotic, such as doxycycline, may be of clinical utility for chronic posterior blepharitis (Table 10-2).
6. Excessive mucous production may be treated with a mucolytic agent, such as acetylcysteine (Table 10-2), compounded to a preparation of 10%, formulated by a pharmacist, for topical ocular use.
7. Topical ocular cyclosporine has been shown to be effective in cases that are refractory to other treatments (Table 10-2). For most severe cases, oral cyclosporine may be used (Table 10-2). However, consultation with an allergist or dermatologist is warranted.
Vernal Keratoconjunctivitis
ICD-9: 372.13—VERNAL CONJUNCTIVITIS
ICD-9: 370.32—LIMBAL AND CORNEAL INVOLVEMENT IN VERNAL CONJUNCTIVITIS
 THE DISEASE
THE DISEASE
VKC is a severe bilateral conjunctival inflammation that affects the pediatric population, usually boys, presenting with symptoms in the first decade of life. In most cases, this condition subsides by the late teens. Unresolved cases, however, develop AKC. VKC is characterized by a peak incidence in the spring, hence the term vernal. Although cases may present year around, symptoms may be mild. More than 90% of patients with VKC have associated atopy (allergic rhinitis, AD, or asthma). VKC may be classified as palpebral disease, limbal disease, or mixed disease.
Pathophysiology
VKC is an immunologic disorder that involves both IgE-mediated and cell-mediated immune mechanisms; thus, type I (immediate) and type IV (delayed) hypersensitivity reactions are involved. Elevated levels of IgE, immunoglobulin G, and mediators of the inflammatory response (histamine, tryptase) have been isolated from patients with VKC. Mast cells, eosinophils, and their mediators play major roles in the development of clinical manifestation of VKC. In addition, Th-2 cells–derived cytokines, chemokines, growth factors, and enzymes are over expressed in the conjunctiva of VKC patients; thus, these factors play a role in the pathophysiology of VKC. The proliferation of fibroblasts within the substantia propia leads to conjunctival thickening. In addition, structural cells such as epithelial cells and fibroblasts are involved both in the inflammatory process and in the tissue remodeling phase, ultimately resulting in the formation of giant papillae (diameter > 1 mm). Corneal changes are likely the result from the effect of inflammatory cells and the release of mediators that are found in high concentrations in the conjunctival epithelium and substantia propria.
Etiology
The etiology of VKC remains unknown.
The Patient
Clinical Symptoms
Patients with VKC complain of intense bilateral itching. In addition, a stringy, mucus discharge is often present. Visual acuity can be affected, especially if patients experience corneal complications or if mucus strands transiently cross the visual axis. Patients may also report foreign body sensation from large papillae or mucus in the tear film. Symptoms typically wax and wane with periods of exacerbation occurring during warmer spring and summer months and decreased symptoms during fall and winter months.
Clinical Signs
Three forms of VKC are generally recognized, palpebral, limbal, and mixed. In the palpebral form, large papillae (cobblestones) develop on the superior tarsal conjunctiva. The papillae are diffuse and may have flattened tops. In the limbal form, gelatinous nodules form in a thickened limbal conjunctiva. These nodules may have small white dots (Horner-Trantas’ dots), which are accumulations of eosinophils. Many patients may present with signs of both limbal and palpebral forms, and hence patients are classified as having mixed VKC. Prominent mucus discharge is often present and can adhere to the giant papillae. In some cases, the discharge can precede the development of giant papillae. The most common corneal finding is punctate keratitis, which can lead to further epithelial breakdown and the development of corneal ulceration or shield ulcer. Shield ulcers are typically located in the superior one third of the cornea, and they tend to have a horizontally oval shape with sharp borders. Shield ulcers are rarely painful. These ulcers are likely the result of epithelial breakdown from exposure to inflammatory mediators or from direct trauma from the giant papillae. Corneal lesions are found in only 3% of severe forms and up to 50% of palpebral forms of VKC. About 30% of patients who develop corneal involvement will have decreased visual acuity. Patients with VKC also have an increased incidence of keratoconus, pellucid marginal degeneration, and cataracts.
Demographics
VKC is a condition that affects younger patients with peak incidence occurring between the ages of 8 and 12. Younger patients tend to be predominately male; however, this sex predilection may be reduced with increasing age of onset. The majority of affected patients are under the age of 20 years. The average duration of VKC is 4 years. In most patients, this condition is resolved by age 30. The disease is more common in dry, warm climates or in areas with polluted air. In more temperate climates, VKC tends to be seasonal with symptoms increasing in the spring and decreasing in the fall. In the sub-Saharan region of Africa, VKC is a significant global health issue.
Significant History
- Personal or family history of atopy (allergic rhinitis, AD, or asthma)
- Medical history of intense itching, lacrimation, foreign body sensation, blepharospasm, and photophobia
Ancillary Tests
VKC remains a clinical diagnosis; therefore, ancillary testing is not done.
The Treatment
1. Supportive ocular treatment: As with AKC, VKC may be treated with the application of cold compresses to the eyelids for 10 minutes, four to six times daily, as well as artificial tears four to six times daily. Maintenance of a cool, moist environment is helpful.
2. Topical ocular corticosteroids: This type of medication is the drug of choice to treat VKC. Up to 85% of patients require its use (Table 10-2).
3. Topical ocular mast cell stabilizers: These medications have an important role in managing chronic cases of VKC (Table 10-2). Clinical improvement may be delayed with mast cell stabilizers; therefore, a topical ocular antihistamine (Table 10-2), antihistamine/mast cell stabilizer (Table 10-2), or corticosteroid (Table 10-2) can be used concomitantly to relieve symptoms. Intractable cases of VKC may respond to a combination of oral aspirin (Table 10-2) and topical ocular cromolyn sodium 2% twice daily.
4. Excessive mucous production may be treated with a mucolytic agent, such as acetylcysteine (Table 10-2), compounded to a preparation of 10%, formulated by a pharmacist, for topical ocular use.
5. Topical ocular cyclosporine has shown to be effective in relieving signs and symptoms of VKC in patients with severe disease (Table 10-2).
6. A short course of low-dose mitomycin-C, compounded to a preparation of 0.01% for topical ocular use, formulated by a pharmacist, may improve signs and symptoms in patients who are intractable to conventional treatment (Table 10-2).
7. Procedural ocular treatment may include supratarsal corticosteroid injection (dexamethasone 2 mg or hydrocortisone 50 mg or triamcinolone 10.5 mg) for refractory giant papillae, surgical excision of giant papillae, cryotherapy of the superior tarsus, and amniotic membrane grafting. Consultation with an oculoplastic or corneal surgeon is recommended.
Giant Papillary Conjunctivitis
ICD-9: 372.30
 THE DISEASE
THE DISEASE
Giant papillary conjunctivitis (GPC) is a severe conjunctival inflammation that occurs most commonly in patients who wear contact lenses. In addition, patients who have ocular prosthesis, exposed sutures, raised corneal scars, filtering blebs, and extruding scleral buckles may develop GPC. This condition represents an immunologic reaction initiated by a prolonged irritation to the superior tarsal conjunctiva. The accumulation of inflammatory cells in the papillae leads to tissue remodeling, resulting in giant papillae that usually have a diameter of at least 1.0 mm.
Pathophysiology
Patients with GPC have an increased number of inflammatory cells (lymphocytes, mast cells, eosinophils, plasma cells, basophils) in the epithelium and substantia propria of the conjunctiva. GPC represents a delayed type IV hypersensitivity reaction. These cells release a variety of inflammatory mediators that act locally, causing vasodilation, edema, and increased mucus production. Structural cells such as epithelial cells and fibroblasts are involved both in the inflammatory process and in tissue remodeling, ultimately resulting in the formation of giant papillae. Patients who develop GPC have high tear film levels of IgE and tryptase, indicating that type I hypersensitivity reaction is involved in the pathology of GPC as well.
Etiology
The etiology of GPC remains unclear. Evidence exists to suggest an immune response is responsible for the tissue changes seen in patients with GPC. Controversy exists over the cause of the inflammatory response. There is also evidence to suggest that the inflammatory response is elicited by an immune reaction to proteins from the lacrimal fluid. These proteins are presumably denatured by lens-hygiene solutions and adhere to the contact lens. Upon insertion of the contact lens, these proteins act as antigens, stimulating the inflammatory response and eventually the development of GPC. In addition to the immune response, mechanical trauma or irritation to the tarsal conjunctiva of the upper lids is also likely to play a role in the development of GPC.
The Patient
Clinical Symptoms
Patients who develop GPC exhibit a predictable set of symptoms. Patients wear the contact lenses without complication for weeks or months, and suddenly symptoms develop. Most cases are bilateral, although unilateral and asymmetric GPC can occur. Patients typically complain of itching, burning, chafing, increased mucus discharge, increased contact lens movement with blinking, decreased contact lens wearing time, and blurring of vision when wearing their contact lenses. In severe cases, patients may complain of eyelid swelling and eyelid drooping. Symptoms generally evanesce when contact lens wear is discontinued.
Clinical Signs
GPC is divided clinically into four stages:
- Stage I. There are no visible changes of the papillae. Upon removal of the contact lens, there may be some minimal mucus and mild itching. Both lens comfort and vision are adequate.
- Stage II. During this stage, the papillae are starting to form and enlarge up to 0.5 mm in diameter. There is a moderate amount of mucus and redness of the palpebral conjunctiva. The contact lens will be mildly coated, and wearing comfort is slightly decreased. Visual acuity through the lens remains adequate.
- Stage III. At this stage, the papillae may have increased to 0.7 mm in diameter. Mucus strands or globs are present, and the contact lens is moderately to heavily coated with a corresponding markedly decreased lens wearing comfort. Vision acuity is variable.
- Stage IV. During this stage, the papillae have a diameter greater than 0.7 mm, and there is a copious amount of mucus discharge resulting in the eyelids sticking together. There is pain on contact lens insertion with excessive movement and often off-center positioning of the lens (high-riding with the lids). Lenses are unwearable, and vision is markedly diminished.
Prominent Signs
- Conjunctival hyperemia
- Mucus discharge causes the eyelids to stick together and limits the mobility of the contact lens during blinking and eye movements
- Giant papillae (cobblestones) of the upper tarsal conjunctiva
- Secondary eyelid edema and ptosis
- Scarring of conjunctiva in advanced cases
- Contact lens coated with a grayish-white film that makes it look dry and dull
Demographics
Most patients with GPC wear soft contact lenses (47.5%), with rigid gas permeable (RGP) contact lens wearers being less prevalent (21.6%). GPC has been reported with ocular prosthesis, exposed sutures from corneal surgery, exposed buckling material from retinal detachment surgery, eyes with corneal deposits of keratin and calcium, band keratopathy, or filtering bleb. Tear film breakup time, the type of ametropia, and keratometry are not risk factors for the development of GPC.
Significant History
- Contact lens wearer
- Improper or poor contact lens hygiene
- History of seasonal allergies
- Younger age
Diagnosis of GPC is primarily clinical.
The Treatment
Stage 1: Refit with frequent replacement or disposable contact lenses, with replacement intervals of 2 weeks to 3 months. Replace conventional hydrogel contact lenses at least two times per year and institute daily cleaning with hydrogen peroxide and unpreserved saline solution. Enzymatic cleaning one to two times per week if contact lenses are replaced at more than 2-week intervals.
Stage 2: Frequent replacement of daily wear contact lenses, with a replacement cycle of 2 to 4 weeks or daily disposable contact lenses. Change contact lens material, that is, from soft hydroxyethyl methacrylate (HEMA) contact lens to non-HEMA material, with replacement every 4 to 6 months. Daily cleaning with hydrogen peroxide and unpreserved saline solution with initiation of a weekly enzymatic cleaning regimen or other designated solution.
Stage 3: Discontinue contact lens wear for approximately 4 weeks. When apical staining or the appearance of tarsal papillae has resolved, refit with frequent replacement or disposable contact lenses worn on a daily wear basis and replaced every 1 to 2 weeks, with daily cleaning with hydrogen peroxide and unpreserved saline solution. An alternative plan is to refit with RGP (high silicon or coated) contact lenses.
Pharmacologic treatment consists of a short pulse course of topical ocular corticosteroid (i.e., loteprednol 0.5%) followed by long-term maintenance with topical ocular mast cell stabilizers (Table 10-2). A topical ocular NSAID may also be effective but may have untoward side effects (Table 10-2).
Stage 4: Discontinue contact lens wear for a minimum of 4 weeks. When apical staining of tarsal papillae and corneal punctuate staining have resolved, refit with frequent replacement or disposable contact lenses worn on a daily wear basis and replaced at 1-week intervals (some patients may need to change contact lenses after 2 to 3 days). Daily cleaning with hydrogen peroxide and unpreserved saline solution will be needed. An alternative plan is to refit with RGP contact lenses. The pharmacologic treatment recommended for Stage 3 may be used for Stage 4.
 FURTHER INFORMATION
FURTHER INFORMATION
www.aad.org American Academy of Dermatology provides access to foundations, institutes, and support groups, plus professional education and resources, news releases, and patient education information pamphlets.
http://online.factsandcomparisons.com Facts & Comparisons 4.0 offers online access to the most up-to-date drug information enabling better therapeutic decisions in less time. Backed by more than 60 years of editorial expertise, you can be assured of unbiased, reliable content from the publishers of the definitive industry reference, Drug Facts and Comparisons.
www.drugs.com Drugs.com is the most popular, comprehensive, and up-to-date source of drug information online, providing free, accurate, and independent advice on more than 24,000 prescription drugs, over-the-counter medicines and natural products.
www.merckmanuals.com As part of its commitment to ensuring that all who need and want medical information can get it, Merck provides the content of these Merck Manuals on the web for free, without any registration, and use is unlimited. The web publications are continuously updated to ensure that the information is as up-to-date as possible.
Juvenile Rheumatoid (Idiopathic) Arthritis
Richard F. Multack, Samuel J. Multack and Leonid Skorin Jr
ICD-9: 714.30
 THE DISEASE
THE DISEASE
Pathophysiology
Juvenile rheumatoid arthritis (JRA), also known as juvenile idiopathic arthritis (JIA), is an autoimmune disease that results in inflammation of the synovia, an oily fluid that lubricates the joints. It is the most common systemic disease associated with iridocyclitis in the pediatric age group. Risk factors for developing uveitis include a positive antinuclear antibody (ANA), being female, and pauciarticular onset. Uveitis will generally develop within 5 years of the onset of the joint disease. The eye may be white with or without pain. The uveitis is often bilateral. Analysis will often reveal a positive ANA, negative rheumatoid factor (RF), and an elevated erythrocyte sedimentation rate (ESR). Based on the mode of onset of the disease and the extent of joint involvement during the first 6 weeks, the following three subgroups of JRA/JIA are recognized:
1. The monoarticular or pauciarticular subgroup involves four or fewer joints and constitutes 40% to 60% of cases. Type 1 is seen in girls less than 5 years of age who have up to a 25% chance of developing chronic iridocyclitis. Type 2 is seen in older boys and has a high association with HLA-B27. These patients are likely to develop acute recurrent iridocyclitis. This subgroup has a relatively high risk of developing uveitis.
2. The polyarticular subgroup affects five or more joints at onset and constitutes 20% to 40% of cases. This subgroup has a moderate risk of developing uveitis (7% to 14%).
3. The systemic onset subgroup results in high remittent fever and at least one of the following features: generalized lymphadenopathy, fever of unknown origin, hepatomegaly, splenomegaly, pericarditis, and a transient maculopapular rash, which is most prominent on the trunk and limbs. This subgroup constitutes 10% to 20% of cases. The risk of developing uveitis in this subgroup is low (<6%).
Etiology
The exact etiology of JRA/JIA is unknown, although various genetic markers have been identified. These include HLA-DR5 and HLA-DPw2. Factors that may contribute are being researched and include infection, a genetic predisposition, an autoimmune response, and hormone interaction.
The Patient
Symptoms and signs, including that of anterior uveitis, depend on which particular subgroup of JRA the patient has. Still’s triad of low-grade uveitis, band keratopathy, and cataracts are found more often in pauciarticular JRA but can be found in any variety. As mentioned earlier, the eye is often white and not inflamed. There may or may not be associated with mild to moderate pain, blurring, or photophobia.
Clinical Symptoms
Most children with JRA present with increased irritability, fatigue, exhaustion, and the reluctance to move a swollen and tender extremity or to walk. The child may also complain of blurred vision or may have a white pupil as seen by the parent or teacher. The anterior uveitis associated with JRA may not cause the patient to seek medical attention. The patient may be asymptomatic, and the eye may not show conjunctival inflammation, but the anterior chamber may be greatly inflamed. Unchecked inflammation can lead to choroidal vascular damage, secondary glaucoma because of the formation of peripheral anterior synechiae and/or posterior synechiae, accelerated cataract formation, macular edema, and phthisis bulbi. A delay in the diagnosis can lead to a poorer prognosis if silent ocular damage is allowed to continue untreated. The long-term outcome will correlate with the severity of the damage present at the time of the initial diagnosis.
Clinical Signs
1. Nonocular signs
- Low-grade fever
- Swollen and tender joint or joints
2. Ocular signs
- Clinically silent in up to 50% of cases
- Nongranulomatous iridocyclitis
- Dyscoria from posterior synechia
- Band keratopathy
- Cataracts—up to 50% of cases
- Secondary glaucoma
- Keratic precipitates (KPs)—small-medium, rarely mutton fat
- Macular edema and macular folds
Demographics
The peak incidence of JRA is between 1 and 4 years of age, but it can appear throughout childhood. The incidence is approximately 113 cases per 100,000 individuals. The ratio of affected females to males is 3:1 with polyarticular involvement, 5:1 in pauciarticular involvement, and equal involvement in the systemic group. The incidence of bilateral ocular involvement varies between 67% and 89%.
Significant History
- Swollen and tender joints
- White pupil
Ancillary Tests
Ocular assessment includes visual acuity, pupil testing, slit-lamp evaluation, tonometry, and dilated fundus examination.
A screening slit-lamp examination should be performed every 3 to 4 months in ANA-positive pauciarticular and polyarticular JRA children whose age of onset was under 7 years. Screenings should be performed every 6 months in pauciarticular or polyarticular JRA children who are ANA negative or whose age of onset was over 7 years of age. Children who develop systemic onset JRA should be examined every 12 months. If no uveitis is discovered after 4 years, the ANA-positive children whose onset was less than 7 years of age can be seen every 6 months, and all patients whose onset was after 7 years of age can be seen annually. After 7 years from the time of onset, all patients are considered to be at low risk for developing uveitis and can be seen yearly from that point onward. Laboratory studies: The incidence of a positive ANA ranges from 71% to 93% in JRA patients with uveitis, compared with 30% in those without uveitis.
The RF is usually negative. The ESR, C-reactive protein (CRP), and immune complexes are useful as nonspecific indicators of disease activity.
The knee is the joint most commonly affected, and a routine knee examination and x-ray can be done in cases where JRA is suspected.
The Treatment
The current trend is to treat even minimal levels of ocular inflammation. It is believed that a chronic, low-grade inflammation can inflict significant damage to the uveal vasculature. More severe and recurrent uveitis requires topical corticosteroids, topical NSAIDS, cycloplegics, periocular injections of depot-steroids, oral nonsteroidal anti-inflammatory agents, oral corticosteroids, immunosuppressive drugs such as low-dose methotrexate, TNF receptor blockers, or immunomodulators such as Abatacept. Systemic immunomodulatory agents may be useful for patients with limited or no response to systemic corticosteroids or those who develop unacceptable adverse effects.
Band keratopathy can be removed by scraping or with chelating agents such as disodium EDTA.
Glaucoma is managed as any secondary form of glaucoma. Standard glaucoma surgeries are often unsuccessful. Antifibrotics (5-fluorouracil, mitomycin-C) and aqueous drainage devices are usually required.
In those patients who develop cataracts, lensectomy with vitrectomy and no intraocular lens placement may be indicated. Prior to cataract extraction, the eye should be quiet for 3 months, the anterior chamber angle should be evaluated for synechiae, and adequate anti-inflammation and immunosuppressive therapy should be used perioperatively.
Pars Planitis
Tammy P. Than
ICD-9: 363.21
 THE DISEASE
THE DISEASE
Pathophysiology
According to the Standardization of Uveitis Nomenclature working group, pars planitis is a chronic inflammation characterized by cells and debris in the vitreous with exudates overlying the peripheral retina, ora serrata, or pars plana ciliaris and occurring in the absence of systemic disease or associated infection. Pars planitis is a more specific clinical entity than intermediate uveitis, which is a broad term used when inflammation is located predominately in the vitreous. Histocompatibility gene typing reveals that HLA-DR15 (suballele of HLA-DR2) is present in 50% to 64% of patients with pars planitis. Increased frequencies of several other HLAs have been noted, including HLA-B8, HLA-B51, HLA-DR17, and HLA-DR2, which suggests an immunogenetic predisposition to pars planitis.
Etiology
The etiology and pathogenesis of pars planitis remain unknown. It may be an autoimmune reaction against the vitreous, peripheral retina, and ciliary body. The differential diagnosis of pars planitis includes intermediate uveitis associated with retinitis pigmentosa, cat scratch disease, Epstein-Barr virus, tuberculosis, Lyme disease, ocular lymphoma, syphilis, and Crohn’s disease. Between 80% and 90% of patients maintain a visual acuity of 20/40 or better in the less involved eye over time.
The Patient
Clinical Symptoms
The symptoms may be so mild that pars planitis may go undetected for several years until young adulthood. The patient may complain of floaters and blurred vision. Pain, photophobia, and ocular redness are usually absent or minimal.
Clinical Signs
The clinical signs, which are bilateral in up to 80% of cases and may wax and wane for 15 to 30 years, include
- Anterior vitreous cells
- Peripheral retinal snowballs (free-floating yellow-white exudates)
- Snowbanks (layered vitreous exudates over the inferior pars plana and ora serrata)
- Variable degree of peripheral retinal periphlebitis
- Absent or mild anterior chamber reaction
Numerous complications result secondary to pars planitis. The three most common are cystoid macular edema (CME), which is usually the cause of decreased vision; cataracts, either secondary to inflammation or corticosteroid usage; and epiretinal membrane formation. Snowbanks are frequently vascularized, which increases the risk of vitreous hemorrhage and may also cause retinal traction leading to a retinal tear and rhegmatogenous retinal detachment. Other possible findings include retinal or choroidal neovascularization, posterior vitreous detachment, optic nerve edema, open angle glaucoma, and neovascular glaucoma. Anterior segment findings include band keratopathy and infrequent posterior synechiae.
Demographics
Pars planitis is a disease of children and young adults that has a bimodal age distribution with peaks at 5 to 15 years and also at 20 to 40 years. The disease rarely presents after age 40. There appears to be a slight predilection for males in the younger group and slightly more females in the older group. The average age of diagnosis is 26 years. Pars planitis accounts for up to 15% of uveitis cases in adults. Prevalence in children is higher, accounting for up to 27% of cases with uveitis.
Significant History
- Insidious onset and may be asymptomatic for several years during childhood
- Increased floaters in younger patient
- Blurred vision in younger patient
Ancillary Tests
Ocular examination requires careful evaluation of the anterior chamber. A significant anterior chamber reaction may be indicative of a panuveitis. A dilated fundus evaluation utilizing binocular indirect ophthalmoscopy with scleral indentation is necessary to visualize snowballs and snowbanks. Fluorescein angiography and optical coherence tomography (OCT) are useful in detecting subtle macular edema and aid in the diagnosis of neovascularization. Although pars planitis is idiopathic, intermediate uveitis is often associated with other diseases; therefore, laboratory testing may include
- CBC with differential
- Syphilis serology (FTA-ABS and RPR)
- Purified protein derivative (PPD)
- Chest radiograph to rule out sarcoidosis and tuberculosis
- Angiotensin-converting enzyme (ACE)
- Lyme titers
- Magnetic resonance imaging (MRI) in patients older than 25 (if other findings and history are suggestive of multiple sclerosis)
The Treatment
Usually, no treatment is administered if acuity is good (20/40 or better). However, because of numerous complications, early aggressive treatment is advocated by some if any amount of macular edema is present.
Oral prednisone, dosed 1 mg/kg of body weight per day, may be prescribed for 2 to 6 weeks followed by a slow taper. Alternatively, periocular corticosteroid injections of methylprednisolone acetate 40 mg or triamcinolone acetonide 40 mg may be administered every 2 to 6 weeks until the resolution of macular edema. Systemic steroids alone manage the inflammation in up to 80% of the cases. Peripheral cryotherapy and peripheral scatter photocoagulation are used to treat neovascularization of the vitreous base. Cryotherapy is not without side effects and has been associated with the development of rhegmatogenous retinal detachments. Pars plana vitrectomy (possibly removing vitreous antigens, inflammatory cells, and inflammatory mediators) may be indicated to remove vitreous opacities or hemorrhages and to reduce or eliminate vitreous traction. If all other treatments prove ineffective, immunosuppressive agents such as methotrexate, azathioprine, chlorambucil, cyclophosphamide, and cyclosporine A may be utilized. Newer therapies such as tacrolimus, etanercept, and infliximab have also proven useful. Combined corticosteroids with immunosuppressant therapy may be warranted in severe cases. Associated macular edema may improve with topical corticosteroid and NSAIDs. Cataract surgery, required in up to 20% of cases, is usually recommended once the eye is inflammation free for 3 months prior to surgery. Topical corticosteroids and cycloplegia are used to manage any secondary anterior chamber reaction. Follow-up is every 1 to 4 weeks during acute phase and every 3 to 6 months during chronic phase. Improvement is slow.
Reactive Arthritis
Tammy P. Than
ICD-9: 099.3—REITER’S DISEASE (SYNDROME)
ICD-9: 372.33—CONJUNCTIVITIS IN MUCOCUTANEOUS DISEASE
 THE DISEASE
THE DISEASE
Pathophysiology
The pathophysiology is uncertain but is thought to be an interaction between bacterial antigens, inflammatory components, and the immune system, combined with a genetic predisposition (HLA-B27). Reactive arthritis is a seronegative spondyloarthropathy encompassing an entire spectrum of disease. Reiter’s syndrome traditionally refers to the classic triad of arthritis, conjunctivitis, and urethritis. A fourth manifestation, mucocutaneous lesions, occurs at high frequency.
Etiology
Reactive arthritis usually begins after an infection of the genitourinary or gastrointestinal tract. The major enteric bacteria are Shigella, Salmonella, Campylobacter, and Yersinia, while the most common venereal infection is Chlamydia trachomatis. The predisposing antigen, HLA-B27, is present in 75% to 90% of cases.
The Patient
Clinical Symptoms
The patient will complain of joint stiffness, heel pain, genitourinary discharge, and skin lesions. Ocular complaints will include mucopurulent discharge, discomfort, red eye, and possibly photophobia.
Clinical Signs
Ocular signs occur in 50% of postvenereal cases and 75% of cases following an enteric infection. Bilateral conjunctivitis is most common and may be observed before or at the onset of arthritis. The second most common ocular finding is acute, nongranulomatous, unilateral anterior uveitis. Ocular complications include cystoid macular edema (CME), epiretinal membrane, optic nerve edema, glaucoma, and cataracts.
Systemic signs include asymmetric arthralgia of the knee, ankle, and toes; sacroiliitis; enthesitis (painful inflammation of tendon or ligament attachment to bone); and talagia (heel pain). The peripheral arthritis is usually acute and migratory. Mucocutaneous lesions are present in 80% of cases and include keratoderma blennorrhagicum (scaling lesions of palms and soles), circinate balanitis (genital ulcers), and painless oral ulcers. Reactive arthritis may also be associated with nail abnormalities (onycholysis, subungual hyperkeratosis), amyloidosis, aortitis, pulmonary fibrosis, prostatitis, and pulmonary fibrosis.
Fewer than 50% of patients present with the complete triad (arthritis, urethritis, and conjunctivitis).
Demographics
The exact incidence is unknown, but European population-based studies estimate it to be 10 to 30/100,000. It is thought to be the most common cause of arthralgia in the 20 to 35 age group. The incidence of reactive arthritis postvenereal disease is more common in men than in women (9:1), with no gender predilection following an enteric infection. This syndrome is more common in whites than African Americans.
Significant History
- History of gastrointestinal disease
- History of genitourinary infection
Ancillary Tests
Ocular evaluation includes a thorough slit-lamp examination of the conjunctiva, cornea, and anterior chamber. A dilated fundus examination is needed to rule out posterior segment complications.
There is no diagnostic test for reactive arthritis, but the following laboratory tests are useful:
- CBC with differential (leukocytosis is common; mild anemia may be present)
- ESR—may be elevated during active stage
- CRP—may be elevated during active stage
- HLA-B27
- Rheumatoid factor—negative
- ANA—negative
- Microimmunofluoresence for C. trachomatis
- Creatinine—elevated
- Radiologic studies corresponding to patients’ arthralgias
The Treatment
Reactive arthritis is often not self-limiting and is recurrent and chronic. Antibiotic treatment has not proven effective in shortening the ocular manifestations. The anterior uveitis should be treated with topical corticosteroids and cycloplegics. If reactive arthritis is secondary to Chlamydial infection, oral azithromycin 1,000 mg for 1 day should be prescribed. The patient’s sexual partners should also be treated. The arthralgia is managed with oral nonsteroidal anti-inflammatories, corticosteroids, or immunosuppressive agents. TNF-α antagonists, such as infliximab, may be beneficial in refractory cases. Enthesitis responds to local corticosteroid injections. The mucocutaneous lesions respond to corticosteroids, phototherapy, and methotrexate. Physical therapy is also useful.
Tuberculosis
Langis Michaud
ICD-9: 017.3
After years of decline, tuberculosis has re-emerged as an important systemic and ocular disease. Being primarily a pulmonary illness, clinical manifestations could also be seen in the eye, brain, intestines, kidneys, and bones.
 THE DISEASE
THE DISEASE
Pathophysiology
Tuberculosis is caused by Mycobacterium tuberculosis, an obligate aerobe that develops in an environment with high oxygen tension, such as the lung apices or in the choroid of the eye. Humans are its only known natural host and the disease is transmitted through direct contact from person to person. Healthy individuals can, once infected, develop a chronic and indolent systemic illness, affecting all tissues or organs of the body. Tuberculosis manifestations vary according to the immune status of the host and the strain of the organism.
There are no exotoxins produced by M. tuberculosis. No endotoxins are found in its cell wall. Primary infection preferentially affects macrophages and other reticuloendothelial cells. An organism that evades destruction by macrophages survives within vacuoles known as phagosomes. M. tuberculosis elicits a chronic inflammatory response, inherent to the control of the infectious process that may lead to extensive tissue damage where T Cells (CD4+ and CD8+) and cytokines interferon gamma (IFN-γ, IL-12, TNF-α, and IL-6) play a major role.
Etiology
Immune reaction to the presence of M. tuberculosis is characterized by a delayed hypersensitivity and an aseptic reaction either to mycobacterial antigens or to the presence of viable mycobacteria invading the tissue, the latter being specifically associated with a TH1 cell–driven granulomatous reaction. Primary ocular tuberculosis occurs when the eye is the initial portal of entry into the body, whereas secondary ocular tuberculosis comes by the spread from an adjacent structure.
The Patient
Ocular Tuberculosis
Most patients with ocular involvement have no history of pulmonary or other systemic forms of tuberculosis. The most common site of infection is the choroid; but, the disease can affect any other ocular structure including the orbit.
Clinical Symptoms
Symptoms vary depending on the ocular structure that is involved. Table 10-3 summarizes possible clinical manifestations of ocular tuberculosis and their associated main symptoms.
TABLE 10-3 Clinical Manifestations of Ocular Tuberculosis
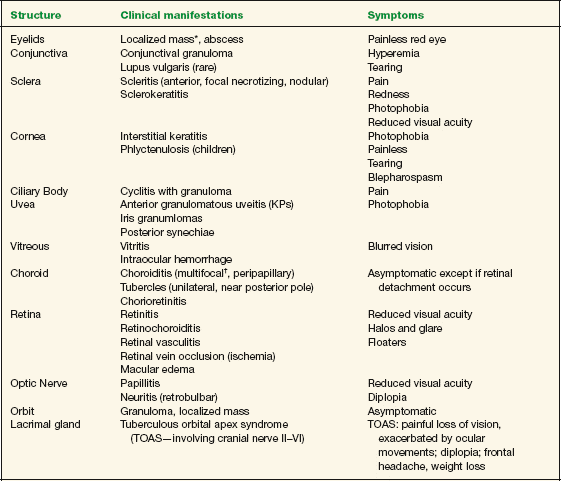
* Primary manifestation (isolated).
† Most frequently encountered sign of tuberculosis involvement of the posterior segment.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


