In this article the epidemiology, pathophysiology, clinical presentation, investigation, management, and prognosis of hypopituitarism and hypothalamic dysfunction, arising from skull base pathologies and treatment of these conditions, are reviewed and discussed. The clinical question: “What is the consequence of pituitary hypofunction in young patients (ie, craniopharyngioma)?” is answered based on information provided in the review.
| EBM Question | Level of Evidence | Grade of Recommendation |
|---|---|---|
| What is consequence of pituitary sacrifice in young patients (ie, craniopharyngioma)? | 1b | A |
The skull base is a complex anatomic region, which forms the floor of the cranial cavity, separating the brain from other facial structures. The ethmoid, sphenoid, occipital, paired frontal, and paired parietal bones divide up the skull base into the anterior, middle, and posterior cranial fossae. Each region contains vital neurovascular structures.
The middle fossa is of particular endocrine importance, as it nests the pituitary gland, or the hypophysis, which is a protrusion from the inferior surface of the hypothalamus. The hypothalamus is sometimes known as the “master clock” and the pituitary the “master gland.” The hypothalamus regulates circadian rhythms by coordinating endogenous biological oscillators with “time giver” signals, such as the light-dark cycle, to keep synchrony with the environment. Through the neurohypophyseal circulation, the hypothalamus communicates with the anterior pituitary gland, which secretes 6 hormones: growth hormone (GH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), adrenocorticotropin (ACTH), thyroid-stimulating hormone (TSH), and prolactin. The posterior pituitary gland secretes anti-diuretic hormone (ADH) and oxytocin.
The hypothalamic-pituitary axis orchestrates a complex neuroendocrine circuitry with other endocrine glands, the principal ones being the thyroid, adrenals, and gonads, to regulate whole body homeostasis. These glands control essential body functions, including appetite, reproductive function, growth and development, energy metabolism, and water balance. Given their central and midline location, the hypothalamus and the pituitary gland are vulnerable to insults and damage by skull base pathologies and radiotherapy targeting the skull base. The most important consequences are hypothalamic dysfunction and hypopituitarism arising from hypothalamic and pituitary gland damage.
Hypothalamic dysfunction can cause disabling obesity, disorders of temperature regulation, or sleep disorders. Hypopituitarism refers to the deficiency of one or more pituitary hormones and results in a range of clinical syndromes with significant morbidity and mortality. It is therefore important to diagnose and manage hypopituitarism early in order to improve both quality of life and survival.
In this review the epidemiology, pathophysiology, clinical presentation, investigation, management, and prognosis of hypopituitarism and hypothalamic dysfunction, arising from skull base pathologies and treatment of these conditions, are discussed. The clinical question “what is the consequence of pituitary hypofunction in young patients (ie, craniopharyngioma)?” is raised and answered.
Hypopituitarism
Epidemiology
Limited information is available on the epidemiology of hypopituitarism in the general population. The prevalence of hypopituitarism was estimated to be 175, 290, and 450 cases per million in 3 cross-sectional surveys. Even less information is available in patients with skull base pathologies, and most are limited to those who have received radiotherapy. Scattered case reports and series describe the development of hypopituitarism in patients following radiotherapy in the 1980s. In these small series, the cumulative probability of varying degrees of hypopituitarism range from 60% to 100%. Pai and colleagues reported a 10-year rate of hypopituitarism in 107 adults following radiotherapy to the skull base, the largest series to date, to be as high as 63%. Samaan and colleagues studied 15 patients who had received radiotherapy for nasopharyngeal cancer, and found hypopituitarism to be present in 14 patients at 20 years’ follow-up.
In the presence of hypopituitarism, mortality is increased. Data from 6 epidemiologic studies report increased mortality, with standardized mortality rates ranging from 1.2 to 2.2. Excess mortality arises from cardiovascular and cerebrovascular diseases. Patients with craniopharyngioma and/or patients treated with radiotherapy appear to be at higher risk.
Pathophysiology
The most common cause of hypopituitarism in patients with skull base pathologies relates to local mass effect or treatment with radiotherapy.
Skull base mass lesions
Neoplasm is the most common skull base mass lesion. Compression of the pituitary stalk portal vessels by the neoplasm is the usual causative mechanism. The lesion can arise from the expanding neoplasm or secondary to an increase in intrasellar pressure. Other mass lesions arise from developmental abnormalities such as craniopharyngiomas, Rathke cleft cysts, and arachnoid cysts. In addition to anterior pituitary hormone deficiencies, craniopharyngiomas can rarely cause diabetes insipidus, due to ADH deficiency.
Craniopharyngiomas are the third most common intracranial tumor, and account for the majority of parapituitary tumors. There is a bimodal peak in incidence at age 5 to 14 years and again after the age of 50. The origin of their development is uncertain. There are frequently large cystic components and the tumors may be intrasellar, extrasellar, or both. Rathke cleft cysts are cystic sellar and suprasellar lesions lined by a single epithelial layer. Arachnoid cysts present at a later age, and are less common than both craniopharyngiomas and Rathke cleft cysts.
Derangement of central endocrine regulation also occurs with other parapituitary space-occupying lesions such as chondromas, suprasellar meningiomas, astrocytomas of the optic nerve, primary tumors of the third ventricle, and secondary metastases.
Radiotherapy
Radiation-induced damage to the hypothalamus and pituitary inevitably occurs when the hypothalamic-pituitary axis lies within the field of radiation. External cranial radiotherapy is commonly used in the treatment of nasopharyngeal carcinomas and parasellar tumors. Deficiency of one or more anterior pituitary hormones is almost invariable following radiotherapy. The most studied patient population has been patients with pituitary tumors treated with radiotherapy. Data are less comprehensive on patients with skull base neoplasms who receive radiotherapy, mainly because of the frequent omission of dynamic testing for GH and ACTH deficiency.
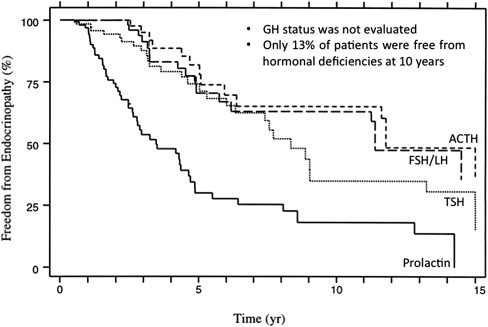
Unlike tumor cells, which are rapidly dividing and are therefore susceptible to nonrepairable “single-hit” radiation damage, the hypothalamic-pituitary axis is more vulnerable to cumulative sublethal radiation. The risk and severity of permanent hypopituitarism varies according to the total dose, the number of fractions, the duration of radiotherapy, and the length of follow-up. The relationship between these parameters and the risk of hypopituitarism was systematically evaluated in 107 adult patients who received radiotherapy (median dose 75 Gy) to the base of skull region for treatment of parasellar tumors. In this study, patients were monitored for up to 10 years (median follow-up 5.5 years) for evidence of hypopituitarism. Fig. 1 shows the Kaplan-Meier estimates of freedom from hypopituitarism following radiotherapy. Five-year actuarial rates of TSH, gonadotropin, and ACTH deficiency were 30%, 29%, and 19%, respectively, which rose to 63%, 36%, and 28% at 10 years. Among the patients who developed hypopituitarism, 47% had more than one axis involved. In a radiation dose analysis, the investigators identified a minimum target dose of 55 Gy as the threshold for pituitary damage, as no patients who received less than 55 Gy to the pituitary gland developed hormone deficiencies. Similar findings were reported in other smaller studies.
Diabetes insipidus from ADH deficiency secondary to radiation is rare and has not been consistently reported. Hayashi and colleagues reported a case series describing impaired ADH secretion in patients who received radiation doses as high as 140 to 180 Gy. Diabetes insipidus was transient, suggesting a much higher dose threshold for radiation-induced diabetes insipidus.
Collectively these studies highlight the importance of endocrine surveillance after radiotherapy to skull base regions, as only approximately 10% of patients were free from hypopituitarism at 10-year follow-up. However, there is one major limitation in these studies: the GH axis is frequently not evaluated. The somatotrophs are the most sensitive to radiation damage. Isolated GH deficiency occurs even after low radiation doses (<30 Gy) to the pituitary gland, and the frequency of GH deficiency rises to 50% to 100% at higher doses. In 2 studies comprising 43 patients with nasopharyngeal carcinomas followed for up to 17 years who underwent the insulin tolerance test (ITT), GH deficiency was diagnosed in up to 90% of patients. The speed of onset of hormonal deficiency is also dependent on the radiation dose. GH deficiency occurs earlier with greater doses. For example, between 2 and 5 years after irradiation, all children receiving more than 30 Gy to the hypothalamic-pituitary axis developed subnormal GH responses to ITT, whereas more than one-third of children who received less than 30 Gy displayed a normal response.
One should be cautious in interpreting the impact of radiation-induced damage to the hypothalamic-pituitary axis on GH status. Depending on the tests used, discordant results may be seen in response to different GH-provocative agents. For example, a normal or mildly insufficient GH response during the combined GH-releasing hormone (GHRH) plus arginine stimulation test was seen in 50% of the patients classified as severely GH deficient using the ITT. The choice and performance of different dynamic tests to evaluate GH sufficiency is discussed in greater detail in the section Investigations.
There is a theoretical advantage in using fractionated stereotactic conformal radiotherapy (SCRT) given the greater precision in irradiation localization, which may reduce radiation damage to normal structures in the brain. However, the incidence of hypopituitarism was not different between conventional radiotherapy and SCRT. Close to a quarter of patients with previously normal pituitary function or partial hypopituitarism who received SCRT developed new hormonal deficit at a median follow-up of 32 months, and panhypopituitarism was seen in 18%.
With the advances in the care in patients with skull base malignancies, survival of these patients has improved. Therefore with increased survival, follow-up evaluation of patients irradiated for tumors of the skull base must focus equally on the possibility of tumor recurrence and the delayed effects of therapy, including the endocrine effects. Endocrine testing should be performed on a yearly basis for at least 10 years and again at 15 years.
Clinical Presentation
The presentation of hypopituitarism is affected by the degree, type, and rate of onset of the pituitary hormone deficiency. Symptoms and signs can be nonspecific and a high index of suspicion is required for diagnosis, which frequently necessitates measurement of basal hormone profile and sometimes dynamic endocrine testing. Radiation-induced hypothalamic-pituitary damage produces a characteristic evolution of pituitary failure of an initial loss of GH secretion, followed by gonadotropins, and finally by failure of ACTH and TSH secretion.
Hypopituitarism arising from radiation-induced damage to the hypothalamic-pituitary axis occurs over years, and the onset of symptoms is typically insidious. The nonspecific symptoms of lethargy, fatigue, low mood, and declining libido are common in cancer survivors and may be difficult to distinguish from symptoms of depression. In addition to cancer-induced cachexia, ACTH deficiency can cause anorexia and weight loss. Table 1 summarizes the symptoms and signs of individual hormone deficiency. The features of isolated deficiencies of each axis are described below.
| Hormone | Symptom | Sign | ||
|---|---|---|---|---|
| Prepubertal | Postpubertal | Prepubertal | Postpubertal | |
| Growth hormone | Growth failure | Loss of vitality, muscle weakness and fatigue, depression | Short stature | Increased fat mass, central adiposity, loss of lean mass, skin thinning and dryness |
| Gonadotropins | Delayed puberty | Loss of libido, sexual dysfunction, infertility | Delayed puberty | Loss of secondary sexual characteristics |
| TSH | Tiredness, cold intolerance, constipation, weight gain, depression, cognitive decline | Puffy face, periorbital edema, coarse skin, loss of eyebrows, delayed relaxation of tendon reflexes | ||
| ACTH | Chronic: Tiredness, fatigue, weight loss, anorexia, myalgia, arthralgia | Hypotension (may be postural only) | ||
| Acute: Presentation can be catastrophic with circulatory shock | ||||
| Prolactin | Failure to lactate | |||
| ADH | Polydypsia, polyuria | Dehydration if intake does not match output | ||
GH deficiency
There has been a recent reappraisal of the role of GH in adult health. While the critical role of GH in stimulating childhood growth is well recognized, GH is the most abundant hormone in the adult pituitary gland and continues to be produced throughout adulthood. GH plays a general role in maintaining critical metabolic processes and the integrity of many tissues. GH deficiency is the earliest manifestation of many forms of hypopituitarism, such as radiation-induced hypothalamic-pituitary damage. However, it frequently remains undiagnosed because of the nonspecificity of the symptoms. Another common reason for overlooking the GH axis is the “myth” that GH replacement is contraindicated in patients with a history of malignancy. There is no convincing evidence for a causal link between GH treatment and tumor recurrence or the development of neoplasia. The relationship between GH replacement and risk of malignancy recurrence is discussed in the section Management.
Before puberty, growth failure is the predominant feature of GH deficiency, which is distinct and rarely missed. Adult-onset GH deficiency is characterized by an impairment of psychological well-being and reduced quality of life. Common symptoms include fatigue, easy exhaustion, and lack of vitality. These patients display dry and atrophic skin, with a disproportionate increase in body fat and concomitant reduction of lean body mass, resulting in significant impairment of physical fitness and muscle strength.
Gonadotropin deficiency
Gonadotropins are important for sexual development, function, and skeletal health. The presentation of hypogonadism is therefore different, depending on the age of individual patients.
In male patients the features are those of delayed puberty, characterized by small penis, small testes, and eunuchoid proportions (span exceeds height >5 cm). Men who have acquired gonadotropin deficiency postpubertally display reduced testicular volume and loss of secondary sexual characteristics, such as loss of facial and body hair and thinning of the skin. Other effects include gynecomastia, erectile dysfunction, and a decrease in skeletal muscle mass, bone mineral density, and general well-being. Azoospermia is found in all cases of hypogonadotropic hypogonadism.
In female patients, gonadotropin deficiency results in primary amenorrhea and absent breast development. In the adult woman, amenorrhea or oligomenorrhea, infertility, breast atrophy, vaginal dryness, and dyspareunia occur. As adrenal androgen production is regulated by ACTH action, a sign of ACTH deficiency is the loss of androgen-dependent pubic and axillary hair.
TSH deficiency
TSH deficiency occurs late in most pituitary disorders. Symptoms are generally milder than in primary hypothyroidism because of some residual TSH secretion. Hypothyroid symptoms include cold intolerance, weight gain, tiredness, and constipation, similar to those seen in primary hypothyroidism. Patients may appear edematous with a pulse at the lower end of the normal range. In severe hypothyroidism, bradycardia and myxedema can occur, although these are rare in patients with TSH deficiency.
ACTH deficiency
ACTH secretion is typically preserved until late in hypopituitarism. However, ACTH deficiency is life-threatening, and patients can present with profound shock if the onset is abrupt. Radiation-induced hypopituitarism usually causes chronic ACTH deficiency. The clinical presentation is characterized by slow and progressive symptoms of fatigue, anorexia, and weight loss. On examination, pallor is common with loss of secondary sexual hair, especially in females.
ADH deficiency
The classic features of diabetes insipidus from ADH deficiency are polydipsia and polyuria with nocturia. ADH deficiency can result in significant dehydration and hypovolemia if fluid intake is not increased to compensate for urinary loss.
Investigations
Diagnosis of hypopituitarism can be difficult, and frequently necessitates referral to an endocrine service with expertise in dynamic testing. Diagnosis in a patient with suspected hypopituitarism begins with history and clinical examination. A high index of suspicion is necessary for clinical diagnosis even among those with suggestive symptomatology, because of the nonspecific nature of symptoms.
In patients with skull base neoplasms who have received radiotherapy, the likelihood of hypothalamic-pituitary dysfunction is sufficiently high to warrant regular endocrine evaluation, even in the absence of typical symptoms. The risk of radiation-induced hypopituitarism is discussed in detail in the section Pathophysiology.
Evaluation of suspected hypopituitarism involves measurement of both baseline and stimulated hormone levels. Evaluation of baseline function includes prolactin, TSH, free thyroxine (T 4 ), cortisol, LH, FSH, and total testosterone in men and estradiol in women. Baseline blood testing reliably identifies hypogonadism, hypothyroidism, and severe hypoadrenalism due to pituitary insufficiency. Table 2 summarizes the endocrine tests for each pituitary hormone deficiency.
| Hormone Deficiency | Screening Test | Confirmatory Test |
|---|---|---|
| Growth hormone | Serum IGF-1 | Insulin tolerance test |
| Gonadotropins | Serum FSH, LH, estrogen (women), total testosterone (men) | Dynamic testing not necessary |
| TSH | Serum TSH, free T 4 | |
| ACTH | Early-morning serum cortisol | Insulin-tolerance test |
| ADH | Paired serum and urine sodium, osmolality | Water-deprivation test |
In postmenopausal women with gonadotropin deficiency, gonadotropin levels are low or undetectable whereas in premenopausal women, low estradiol levels and low or normal gonadotropin levels, with a clinical presentation of amenorrhea or oligomenorrhea, are sufficient evidence of the diagnosis. In adult men, a similar picture of low testosterone levels and low or inappropriately normal gonadotropin levels is seen.
The biochemical picture of secondary hypothyroidism is similar to that seen in secondary hypogonadism. Free T 4 concentration is low, in association with a low or inappropriately normal serum TSH concentration.
Serum insulin-like growth factor 1 (IGF-1) concentration may be measured for evaluation of GH deficiency, but is only useful when age-adjusted normal ranges are used. While a low age-adjusted IGF-1 level is seen in adult GH deficiency, a normal concentration does not exclude the diagnosis. However, a subnormal IGF-1 level in an adult patient with coexisting pituitary hormone deficits is strongly suggestive of GH deficiency, particularly in the absence of conditions known to reduce IGF-1 levels such as malnutrition, liver disease, poorly controlled diabetes mellitus, and hypothyroidism. Patients with 3 or more pituitary hormone deficiencies and an IGF-1 level below the reference range have a greater than 97% chance of being GH deficient, and therefore do not require a GH stimulation test.
Dynamic Testing
Provocative endocrine testing is indicated for those with suspected ACTH and GH deficiency, as basal hormonal profile does not distinguish those with mild to moderate deficiency from normal individuals. The ITT is the test of choice, as it evaluates for GH and ACTH deficiency concurrently. Following injection of a standard dose of intravenous insulin (0.1 unit/kg), GH and cortisol concentrations are serially measured. The ITT evaluates the response of the hypothalamic-pituitary-adrenal axis to the potent stressor of hypoglycemia, and is generally the gold standard in the confirmation of GH deficiency and secondary adrenal failure.
GH deficiency
Several provocative tests are available for the testing of GH deficiency. The ITT was recommended as the test of choice by the Growth Hormone Research Society. Although the GHRH plus arginine test and the GHRH plus GH-releasing peptide test are valid alternatives to the ITT, both of these directly stimulate GH release from the pituitary gland and may miss GH deficiency due to hypothalamic disease. This finding is particularly relevant to radiation-induced hypothalamic-pituitary dysfunction, in which the ITT shows the greatest sensitivity and specificity within the first 5 years after irradiation, whereas false negatives may occur when GHRH-based tests are used.
The ITT, which should be performed in experienced endocrine units under supervision, distinguishes GH deficiency from the reduced GH secretion that accompanies normal aging. Severe GH deficiency is defined by a peak GH response to hypoglycemia of less than 3 μg/L.
ACTH deficiency
The highest plasma cortisol levels are found between 6:00 am and 8:00 am in normal individuals, and the lowest before midnight. If an early-morning cortisol level is less than 100 nmol/L, cortisol deficiency is highly likely, whereas a baseline level greater than 500 nmol/L indicates normality and thus dynamic testing may not be required.
For patients with intermediate cortisol levels, ITT is indicated. On achievement of adequate hypoglycemia (<2.2 mmol/L), a peak cortisol response of between 500 and 600 nmol/L is generally accepted as adequate.
The short Synacthen (tetracosactrin) test is not useful in the evaluation of pituitary reserve. An exogenous bolus of synthetic ACTH is a good test of adrenal reserve, but does not reliably detect ACTH deficiency, as in the case of radiation-induced hypopituitarism.
ADH deficiency
The diagnosis of ADH deficiency is suggested by the presence of polyuria (more than 40 mL/kg per 24 hours). To confirm the diagnosis, an 8-hour fluid-deprivation test is required, which should be performed under strict observation in an endocrine center, because of the risk of severe fluid and electrolyte depletion. In patients with ADH deficiency the urine fails to concentrate, resulting in a rapid increase in plasma osmolality.
Management
Endocrine replacement therapy to restore normal physiologic circulating hormone levels is the goal in hypopituitarism treatment. The aim is to mimic the normal hormonal milieu as far as possible, thus improving symptoms while avoiding overtreatment. The form and dose of hormone required depend on the physiologic need of each individual, which varies with age, gender, and health status. Endocrine replacement therapy for each anterior pituitary hormone is discussed in this section.
GH replacement
GH replacement stimulates protein synthesis, lipolysis, and fat oxidation, and reduces protein oxidation, which results in an increase in lean body mass and a decrease in fat mass. Bone mineral density showed progressive increase up to 5%, with the level being twice as high in GH-treated subjects as in controls. GH replacement also improved perceived health status, vitality, mood, and subjective well-being, which are frequent symptoms in patients with skull base malignancies.
There has been concern that GH may increase the risk of tumor recurrence, given that GH promotes tissue growth. However, this concern is theoretical with no convincing supportive evidence. This risk of tumor progression or recurrence is unproven. In the Hypopituitary Control and Complications Study Italian Database, 90 patients with craniopharyngioma were treated with GH with no evidence of tumor regrowth. There was no relationship between dose of GH and treatment modalities in more than 1000 patients with craniopharyngioma treated with GH for 10 years. Chung and colleagues prospectively followed 50 patients with a range of parasellar tumors in addition to craniopharyngioma, including germ cell tumor, arachnoid cyst, meningioma, glioma, and mesenchymal tumor, for 3 years, and GH replacement was abandoned in only 1 patient because of an apparent increase in tumor volume. In addition, overall cancer incidence rates in acromegalic patients were lower than those in the general population. Collectively these data provide strong evidence against a causal association between GH replacement and an increased risk of malignancy.
GH replacement should be initiated and monitored by an endocrinologist with experience in the treatment of hypopituitarism. The starting dose of GH in young men and women is 0.2 and 0.3 mg/d, respectively, and in older individuals 0.1 mg/d. Subsequent monitoring of serum GH and IGF-1 levels guide dose escalation, which should be individualized and guided by clinical response.
The most common side effect of GH is fluid retention, which can manifest as edema, paresthesia, and carpal tunnel syndrome. The effects are usually mild and resolve either spontaneously or with dosage reduction.
Sex-steroid replacement
Sex-steroid replacement maintains normal body composition, skeletal health, and sexual function in hypogonadal patients. It is therefore the most appropriate form of replacement therapy. Gonadotropin therapy is indicated only in patients planning pregnancy.
In women, estrogen is provided by many standard hormone-replacement therapies, together with progesterone, the latter in women with an intact uterus. A nonoral route (eg, transdermal, gel, or implant) is recommended because oral estrogen reduces hepatic IGF-1 production and worsens the metabolic impact of GH deficiency.
For men various formulations of androgen replacement are available, and the choice of preparation depends on availability and patient preference. Recently, testosterone undecanoate, a long-acting intramuscular injection, has become available, providing stable serum testosterone levels for more than 3 months. This new preparation may therefore be more convenient than traditional short-acting intramuscular formulations and implants.
ACTH deficiency
Glucocorticoid requirement increases during acute illness. The goal of glucocorticoid replacement is to provide physiologic levels but to ensure sufficiency during times of acute illness. Different formulations are available, including hydrocortisone, cortisone acetate, prednisolone, and dexamethasone, with differing pharmacokinetics and pharmacodynamics. In general, the lowest replacement dose tolerated by the patient is preferred (equivalent to 10–20 mg cortisol per day). Doses should be divided to suit individual needs and overreplacement should be avoided, as it may worsen metabolic profile.
It is of paramount importance that any patient identified as having ACTH deficiency should be educated about its clinical implications. An appropriate Medic-alert bracelet or necklace should be worn, and patients should be instructed to increase the replacement dose twofold to threefold in the case of an intercurrent illness or when undergoing surgery.
TSH deficiency
T 4 -replacement therapy is the treatment of choice for TSH deficiency, and patients are treated in the same way as in primary hypothyroidism. The normal starting dose in a young patient without evidence of cardiac disease is 1.5 μg/kg/d. In a patient suspected to have hypopituitarism, T 4 therapy should be delayed until ACTH deficiency has been excluded or treated, as T 4 replacement may worsen cortisol deficiency if present. Measurement of TSH is unhelpful in the monitoring of T 4 -replacement therapy in secondary hypothyroidism, and thyroxine dosage should be titrated according to serum free T 4 level.
ADH deficiency
Desmopressin is the drug of choice for the treatment of diabetes insipidus, and can be administered orally or intranasally. Desmopressin is commenced at a low dose and the dose increased according to clinical response. The aim is to control urine output, and the dose required can vary considerably between different patients. Serum sodium level should be monitored to prevent development of hyponatremia from overreplacement.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


