Purpose
To assess the diagnostic yield and the practicality of implementing whole exome sequencing within a clinical ophthalmology setting.
Design
Evaluation of a diagnostic protocol.
Methods
setting : Patient participants were enrolled during clinical appointments in a university-based ophthalmic genetics clinic. patient population : Twenty-six patients with a variety of presumed hereditary retinal dystrophies. intervention : Participants were offered whole exome sequencing in addition to clinically available sequencing gene panels between July 2012 and January 2013 to determine the molecular etiology of their retinal dystrophy. main outcome measures : Diagnostic yield and acceptability of whole exome sequencing in patients with retinal disorders.
Results
Twenty-six of 29 eligible patients (∼90%) who were approached opted to undergo molecular testing. Each participant chose whole exome sequencing in addition to, or in lieu of, clinically available sequencing gene panels. Time to obtain informed consent was manageable in the clinical context. Whole exome sequencing successfully identified known pathogenic mutations or suspected deleterious variants in 57.7% of participants. Additionally, 1 participant had 2 autosomal dominant medically actionable incidental findings (unrelated to retinopathy) that were reported to enable the participant to take preventive action and reduce risk for future disease.
Conclusions
In this study, we identified the molecular etiology for more than half of all participants. Additionally, we found that participants were widely accepting of whole exome sequencing and the possibility of being informed about medically actionable incidental findings.
The pace of progress in ophthalmic genetics has been exponential over the last decade. It is critical for ophthalmologists to understand emerging diagnostic technologies that may have clinical implications for their patients in the very near future. Whole exome sequencing (exome sequencing) and massively parallel sequencing gene panels are attractive new testing approaches for diagnosing genetic disorders that exhibit genetic heterogeneity and overlapping phenotypes. Few Mendelian disorders exhibit the degree of genetic heterogeneity demonstrated by retinitis pigmentosa (RP), one of the most common retinal dystrophies. Over 100 genes have been associated with this condition, yet only half of all patients with RP have an identifiable mutation. Moreover, other retinal dystrophies, including cone-rod dystrophy, cone dystrophy, and Stargardt disease, also exhibit genetic heterogeneity. Further complicating the clinical assessment of these disorders is the fact that retinal disorders also demonstrate significant phenotypic heterogeneity. For instance, mutations in the ABCA4 gene have been associated with several hereditary retinal dystrophies (Stargardt disease, cone-rod dystrophy, cone dystrophy, and RP).
Prior to the advent of massively parallel sequencing, genetic testing for heterogeneous disorders was pursued 1 gene at a time or through limited and expensive gene panels via Sanger sequencing. The benefit of using a broader testing methodology in such circumstances is the potential to eliminate the guesswork inherent in choosing only a subset of genes to test. Another advantage is that many participants seen in ophthalmic genetics clinics report no family history of retinal dystrophy, complicating determinations of inheritance patterns that might otherwise guide diagnostic strategies. Since retinal dystrophies are thought to be almost exclusively hereditary in nature, one can assume that there are yet-unidentified genes associated with RP and other retinal dystrophies. Exome sequencing allows the clinician the flexibility of ordering a single test for all suspected heterogeneous disorders and allows the laboratory the flexibility to analyze newly reported genes without continuously updating testing platforms.
A potential complication of exome sequencing testing vs targeted massively parallel sequencing gene panels is the prospect of patients receiving incidental findings unrelated to the retinal diagnosis. That is, when essentially all genes in an individual’s genome are sequenced, information will be potentially available regarding other genetic disorders unrelated to the indication for testing. The American College of Medical Genetics (ACMG) recommends that laboratories return selected medically actionable incidental findings as part of any genome-scale clinical test. Thus, a small but predictable subset of patients will have such additional findings. It is uncertain how patients might react to this possibility in the clinical setting; therefore, additional time is required to discuss the likelihood and examples of incidental findings as part of the informed consent process.
The goals of this study were to investigate the use of exome sequencing to identify the molecular etiology of retinal dystrophies in a clinical ophthalmology setting and to determine the feasibility of using this novel and complex form of genetic testing, with regard to the potential discovery of incidental findings. Previous studies have shown the effectiveness of exome sequencing and targeted gene panels in determining the molecular etiology of retinal dystrophies. Here we demonstrate its high diagnostic yield, feasibility, and acceptability of exome sequencing for retinal dystrophy patients enrolled in a clinical setting.
Subjects and Methods
Patients evaluated for retinal disorders in the University of North Carolina Kittner Eye Center Ophthalmic Genetics Clinic between July 2012 and January 2013 were invited to participate. Participants were enrolled in the research protocol to undergo research genetic testing during their initial or follow-up clinical visits. Return patients were eligible if the molecular etiology of their retinal disorder was unknown. All potential participants were offered clinically available massively parallel sequencing targeted gene panel testing and research exome sequencing through this study. The University of North Carolina at Chapel Hill Institutional Review Board approval was obtained prior to patient enrollment, and this study adhered to the tenets of the Declaration of Helsinki.
All participants were enrolled and consented by a certified genetic counselor and agreed to learn of any diagnostic-related findings as well as any medically actionable incidental findings. Known pathogenic mutations and variants of unknown clinical significance that could potentially explain their retinal disease were returned to participants. However, only clearly pathogenic medically actionable incidental findings were returned. Thus, variants of unknown significance within genes associated with medically actionable findings were not returned to participants given their uncertainty and low a priori risk of being pathogenic in presumably unaffected individuals. The list of conditions in the category of medically actionable incidental findings was based on a schema previously described by our group and further refined by a committee of medical and molecular geneticists, genetic counselors, a neurologist, a cardiologist, and an ethicist as part of the NCGENES Study currently being conducted at University of North Carolina at Chapel Hill. This list included all conditions recently recommended by the American College of Medical Genetics for return of incidental findings.
Exome sequencing was performed using Agilent’s SureSelect XT Target Enrichment System (Agilent Technologies, Santa Clara, California) for Illumina (Illumina, Inc., San Diego, California) paired-end sequencing on the HiSeq 2000 instrument. The average depth of coverage for all participants across the entire region targeted for enrichment was 58.19.
We used a custom pipeline developed for the NCGENES project to process raw sequence data from FASTQ files to generate variant calls. Variants were stored in a database and extensively annotated.
To facilitate evaluation of variants possibly related to the participants’ retinal disorder, we filtered the exome data using a comprehensive list of 186 genes previously associated with syndromic and nonsyndromic retinopathies, which was curated using Online Mendelian Inheritance of Man (OMIM), GeneTests.org , relevant medical literature, and genes currently being evaluated in clinical laboratories. Participants’ exome data were reanalyzed using an updated gene list, including 214 genes, 1 year later. A complete list of these genes is available in Supplemental Table 1 (Supplemental Material available at AJO.com ). Variants within this set of genes were then prioritized into computational classes by an informatics algorithm to select: (1) variants previously reported as mutations in the Human Gene Mutation Database ; (2) predicted truncating variants that demonstrated <1% minor allele frequency; (3) missense variants with <1% minor allele frequency; and several other categories with decreasingly likely pathogenicity. Variants were then analyzed for pathogenicity using a custom user interface. The total number of exome and filtered variants for each participant is available in Supplemental Table 2 (Supplemental Material available at AJO.com ). Manual analysis of filtered variants entailed a combination of literature searches, publicly available variant databases queries, locus-specific database searches, Condel in silico modeling, and evolutionary conservation.
The veracity of potential disease-causing variants identified by exome sequencing and passing manual curation were confirmed by Sanger sequencing on a duplicate sample in the CLIA-certified University of North Carolina McLendon Molecular Genetics Laboratory. All participants were asked to return for a follow-up research appointment to discuss results and were provided with a research report summarizing the yield of the exome sequencing analysis. Participants were not given the option of learning about non–medically actionable incidental results.
Results
During the enrollment period, 29 patients were eligible (15 new and 14 return clinic patients). Twenty-six of the 29 patients (∼90%) opted to undergo clinically available testing and/or research exome sequencing. Three new patients declined both clinical and research testing. All 26 patients who wished to proceed with genetic testing chose exome sequencing, and 3 of the new patients (Participants 11, 20, and 25) opted to have simultaneous clinically available genetic testing. Participant 11 had negative clinical testing and Participants 20 and 25 had pathogenic variants identified that were also detected via exome sequencing.
The informed consent process for return patients opting for exome sequencing took 30 minutes or less. New patient appointments lasted 75 minutes, and the informed consent process took approximately 30 minutes. All questions about exome sequencing were addressed, and none of the participants shared any concerns regarding the possibility of learning medically actionable incidental findings.
Participants represented a wide variety of ages and clinical indications, outlined in Table 1 . Five of 26 participants (∼19%) were under 18 years of age, and the adults ranged in age from 22 to 69 years. The majority of participants had a firm clinical diagnosis; however, 3 of 26 participants (∼11%) had an uncertain clinical diagnosis at the time of enrollment. Fourteen of 26 participants (∼54%) did not have a known family history of retinal dystrophy.
| Participant | Age at Enrollment (y) | Clinical Diagnosis | Age of Onset | Family History | Previous Testing Results |
|---|---|---|---|---|---|
| 1 | 34 | Retinitis pigmentosa | Early teens | No | N/A |
| 2 | 60 | Cone dystrophy | 20s | No | N/A |
| 3 | 9 | Retinal dystrophy NOS + hearing loss | Hearing loss – congenital Retinal dystrophy – ∼5 | Yes – AD | N/A |
| 4 | 46 | Cone-rod dystrophy | Early 20s | No | N/A |
| 5 | 48 | Cone dystrophy | Congenital | Yes – XL | N/A |
| 6 | 60 | Retinitis pigmentosa | 29 years | No | N/A |
| 7 | 26 | Stargardt disease | 16 years | Yes – AR | N/A |
| 8 | 43 | Retinal dystrophy NOS | 41 years | No | N/A |
| 9 | 23 | Macular dystrophy | 10 years | No | N/A |
| 10 | 41 | Stargardt disease | 6 years | No | ABCA4 c.161G>A (p.Cys54Tyr) |
| 11 | 7 | Familial exudative vitreoretinopathy | ∼6 years | Yes – AD | Negative 4 adFEVR panel + NDP gene |
| 12 | 68 | Retinitis pigmentosa | ∼62 years | No | N/A |
| 13 | 52 | Cone-rod dystrophy | ∼42 years | No | N/A |
| 14 | 64 | Retinitis pigmentosa | 34 years | Yes – AR | N/A |
| 15 | 22 | Retinitis pigmentosa | 15 years | No | N/A |
| 16 | 12 | Retinal dystrophy NOS | 11 years | No | Negative 13 recessive retinitis pigmentosa gene panel |
| 17 | 48 | Retinitis pigmentosa | ∼38 years | No | N/A |
| 18 | 23 | Usher syndrome type 2 | Hearing loss – 1 year retinitis pigmentosa – 13 years | Yes – AR | N/A |
| 19 | 36 | Retinitis pigmentosa | 35 years | No | N/A |
| 20 | 30 | Retinitis pigmentosa | 30 years | Yes – AD | RP1 c.2029C>T (p.R677X) |
| 21 | 26 | Leber congenital amaurosis | Congenital | Yes – AD | N/A |
| 22 | 29 | Retinitis pigmentosa | 15 years | Yes – AD | Negative RHO , RDS , RP1 (c.1500-3200), PRPF31 , PRPF8 (exon 42), PRPF3 (exon11), NR2E3 (c.150-210), TOPORS (c.1975-2820), IMPDH1 (exon 10), RP2 , RPGR , SNRNP200 |
| 23 | 9 | Retinitis pigmentosa | 6 years | Yes – AD | N/A |
| 24 | 54 | Stargardt disease | Mid-30s | Yes – AR | N/A |
| 25 | 11 | Retinitis pigmentosa | 9 years | Yes – XL | RPGR c.2323_2324delGA (p.Arg775fs) |
| 26 | 44 | Retinitis pigmentosa | ∼42 years | No | N/A |
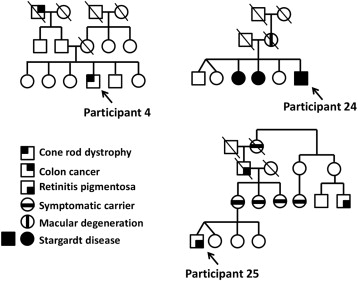
Fifteen of 26 participants (∼58%) had clearly deleterious mutations or highly suspicious variants of unknown significance consistent with their phenotype. A complete summary of previous genetic testing and exome sequencing results for each participant is shown in Table 2 . Each of these variants was confirmed using Sanger sequencing. Participant 4 had 2 different medically actionable incidental findings: an apparently novel nonsense variant in BRCA2 , c.2857G>T (p.Glu593*), which is associated with hereditary breast ovarian cancer syndrome; and an apparently novel frameshift variant in MSH6 , c.1170delT (p.Phe391fs), which is associated with Lynch syndrome. Both truncating variants were considered likely pathogenic, given their suspected effect on the protein. When questioned further about family history of cancer at results disclosure, he reported that his paternal grandfather had a history of colon cancer at an older age ( Figure ).
| Participant | Exome sequencing Diagnostic Results | Previous Reports | Minor Allele Frequency a | Conservation | Condel Score b | Interpretation | |
|---|---|---|---|---|---|---|---|
| 1 | NC_000003.11:g.129252554C>T | RHO c.1040C>T (p.Pro347Leu) | Pathogenic | 8.263e-06 | Highly conserved | 0.698 Deleterious | Known pathogenic |
| 2 | Negative | ||||||
| 3 | NC_000019.9:g.48343048G>A | CRX c.724G>A (p.Val242Met) c | Pathogenic | 0.001435 | Not conserved | 0.853 Deleterious | Likely benign |
| 4 | NC_000001.10:g.94528806A>G | ABCA4 c.1622T>C (p.Leu541Pro) | Pathogenic | 0.0001235 | Highly conserved | 1.000 Deleterious | Known pathogenic |
| NC_000001.10:g.94528265C>T | ABCA4 c.1805G>A (p.Arg602Gln) | Pathogenic | 1.68e-05 | Highly conserved | 0.451 Deleterious | Known pathogenic | |
| NC_000001.10:g.94508969G>A | ABCA4 c.3113C>T (p.Ala1038Val) | Polymorphic Pathogenic | 0.001426 | Mostly conserved | 0.000 Neutral | Uncertain variant | |
| NC_000013.10:g.32911349G>T | BRCA2 c.2857G>T (p.Glu593*) | None | 0 | — | — | Likely pathogenic | |
| NC_000002.11:g.48026292_48026292delT | MSH6 c.1170delT (p.Phe391fs) | None | 0 | — | — | Likely pathogenic | |
| 5 | Negative | ||||||
| 6 | Negative | ||||||
| 7 | NC_000001.10:g.94466624C>T | ABCA4 c.6320G>A (p.Arg2107His) | Pathogenic | 0.001872 | Highly conserved | 1.000 Deleterious | Known pathogenic |
| NC_000001.10:g.94496666G>A | ABCA4 c.4139C>T (p.Pro1380Leu) | Pathogenic | 0.0002012 | Highly conserved | 0.619 Deleterious | Known pathogenic | |
| NC_000001.10:g.94520708A>G | ABCA4 c.2546T>C (p.Val849Ala) | Pathogenic | 0.001335 | Not conserved | 0.007 Neutral | Uncertain variant | |
| 8 | Negative | ||||||
| 9 | NC_000007.13:g.33380565C>G | BBS9 c.1255C>G (p.Pro419Ala) | None | 9.078e-05 | Mostly conserved | 0.026 Neutral | Uncertain variant |
| NC_000007.13:g.33545217A>T | BBS9 c.2138A>T (p.Glu713Val) | None | 0 | Mostly conserved | 0.922 Deleterious | Uncertain variant | |
| 10 | NC_000001.10:g.94577135C>T | ABCA4 c.161G>A (p.Cys54Tyr) | Pathogenic | 1.65e-05 | Mostly conserved | 0.026 Neutral | Uncertain variant |
| NC_000004.11:g.16008270C>T | PROM1 c.1345G>A (p.Val449Met) | None | 0.001475 | Mostly conserved | 0.810 Deleterious | Likely benign d | |
| 11 | Negative | ||||||
| 12 | NC_000001.10:g.216108128T>C | USH2A c.7130A>G (p.Asn2377Ser) | VUS Polymorphism | 0.003772 | Mostly conserved | 0.001 Neutral | Uncertain variant |
| NC_000001.10:g.215901511G>A | USH2A c.11927C>T (p.Thr3976Met) | VUS | 0.0005192 | Highly conserved | 0.966 Deleterious | Uncertain variant | |
| 13 | Negative | ||||||
| 14 | Negative | ||||||
| 15 | NC_000011.9:g.66293652T>G | BBS1 c.1169T>G (p.Met390Arg) – Homozygous | Pathogenic | 0.001484 | Highly conserved | 0.627 Deleterious | Known pathogenic |
| 16 | NC_000014.8:g.68195946G>C | RDH12 c.697G>C (p.Val233Leu) | Pathogenic | 2.491e-05 | Highly conserved | 0.910 Deleterious | Uncertain variant |
| NC_000014.8:g.68200483T>G | RDH12 c.869T>G (p.Val290Gly) | None | 8.246e-05 | Highly conserved | 0.930 Deleterious | Uncertain variant | |
| 17 | Negative | ||||||
| 18 | NC_000001.10:g.216498866_216498867insTGGC | USH2A c.923_924insGCCa (p.His308fs) | Pathogenic | 0.0001074 | — | — | Known pathogenic |
| NC_000001.10:g.215853632_215853633insAA | USH2A c.12152_12153insTT (p.Glu4051fs) | None | 0 | — | — | Likely pathogenic | |
| 19 | NC_000001.10:g.216420460C>A | USH2A c.2276G>T (p.Cys759Phe) | Pathogenic | 0.000784 | Highly conserved | 1.00 Deleterious | Known pathogenic |
| NC_000001.10:g.215848961A>T | USH2A c.12295-3T>A | None | — | — | Uncertain variant | ||
| 20 | NC_000008.10:g.55538471C>T | RP1 c.2029C>T (p.R677X) | Pathogenic | 0 | — | — | Known pathogenic |
| 21 | Negative | ||||||
| 22 | NC_000007.13:g.128034615C>T | MERTK c.1787-2A>C Apparently homozygous e | None | 0 | — | — | Likely pathogenic |
| 23 | Negative | ||||||
| 24 | NC_000006.11:g.42689616T>C | PRPH2 c.457A>G (p.Lys153Glu) | None | 0 | Highly conserved | 0.873 Deleterious | Uncertain variant |
| 25 | NC_000023.10:g.38145928_38145929delCT | RPGR c.2323_2324delGA (p.Arg775fs) | Pathogenic | 0 | — | — | Known pathogenic |
| 26 | NC_000001.10:g.94528143C>T | ABCA4 c.1927G>A (p.Val643Met) | Pathogenic | 0.001618 | Highly conserved | 0.383 Neutral | Uncertain variant |
a Minor allele frequencies were obtained using the Exome Aggregation Consortium (ExAC) Browser.
b The Condel score is provided as a reference only and was not taken into consideration in variant interpretation.
c Not confirmed by Sanger sequencing owing to variant being likely benign and not reported back to participant.
d Parental studies by Sanger sequencing identified both the ABCA4 and PROM1 variants in the unaffected father, suggesting that the PROM1 variant was likely benign.
e An ad hoc analysis examining coverage data suggests that this patient has a partial deletion of the MERTK gene in lieu of being homozygous for the splice variant. The participant’s parents are deceased. Therefore, we are unable to perform parental studies.
Results
During the enrollment period, 29 patients were eligible (15 new and 14 return clinic patients). Twenty-six of the 29 patients (∼90%) opted to undergo clinically available testing and/or research exome sequencing. Three new patients declined both clinical and research testing. All 26 patients who wished to proceed with genetic testing chose exome sequencing, and 3 of the new patients (Participants 11, 20, and 25) opted to have simultaneous clinically available genetic testing. Participant 11 had negative clinical testing and Participants 20 and 25 had pathogenic variants identified that were also detected via exome sequencing.
The informed consent process for return patients opting for exome sequencing took 30 minutes or less. New patient appointments lasted 75 minutes, and the informed consent process took approximately 30 minutes. All questions about exome sequencing were addressed, and none of the participants shared any concerns regarding the possibility of learning medically actionable incidental findings.
Participants represented a wide variety of ages and clinical indications, outlined in Table 1 . Five of 26 participants (∼19%) were under 18 years of age, and the adults ranged in age from 22 to 69 years. The majority of participants had a firm clinical diagnosis; however, 3 of 26 participants (∼11%) had an uncertain clinical diagnosis at the time of enrollment. Fourteen of 26 participants (∼54%) did not have a known family history of retinal dystrophy.
| Participant | Age at Enrollment (y) | Clinical Diagnosis | Age of Onset | Family History | Previous Testing Results |
|---|---|---|---|---|---|
| 1 | 34 | Retinitis pigmentosa | Early teens | No | N/A |
| 2 | 60 | Cone dystrophy | 20s | No | N/A |
| 3 | 9 | Retinal dystrophy NOS + hearing loss | Hearing loss – congenital Retinal dystrophy – ∼5 | Yes – AD | N/A |
| 4 | 46 | Cone-rod dystrophy | Early 20s | No | N/A |
| 5 | 48 | Cone dystrophy | Congenital | Yes – XL | N/A |
| 6 | 60 | Retinitis pigmentosa | 29 years | No | N/A |
| 7 | 26 | Stargardt disease | 16 years | Yes – AR | N/A |
| 8 | 43 | Retinal dystrophy NOS | 41 years | No | N/A |
| 9 | 23 | Macular dystrophy | 10 years | No | N/A |
| 10 | 41 | Stargardt disease | 6 years | No | ABCA4 c.161G>A (p.Cys54Tyr) |
| 11 | 7 | Familial exudative vitreoretinopathy | ∼6 years | Yes – AD | Negative 4 adFEVR panel + NDP gene |
| 12 | 68 | Retinitis pigmentosa | ∼62 years | No | N/A |
| 13 | 52 | Cone-rod dystrophy | ∼42 years | No | N/A |
| 14 | 64 | Retinitis pigmentosa | 34 years | Yes – AR | N/A |
| 15 | 22 | Retinitis pigmentosa | 15 years | No | N/A |
| 16 | 12 | Retinal dystrophy NOS | 11 years | No | Negative 13 recessive retinitis pigmentosa gene panel |
| 17 | 48 | Retinitis pigmentosa | ∼38 years | No | N/A |
| 18 | 23 | Usher syndrome type 2 | Hearing loss – 1 year retinitis pigmentosa – 13 years | Yes – AR | N/A |
| 19 | 36 | Retinitis pigmentosa | 35 years | No | N/A |
| 20 | 30 | Retinitis pigmentosa | 30 years | Yes – AD | RP1 c.2029C>T (p.R677X) |
| 21 | 26 | Leber congenital amaurosis | Congenital | Yes – AD | N/A |
| 22 | 29 | Retinitis pigmentosa | 15 years | Yes – AD | Negative RHO , RDS , RP1 (c.1500-3200), PRPF31 , PRPF8 (exon 42), PRPF3 (exon11), NR2E3 (c.150-210), TOPORS (c.1975-2820), IMPDH1 (exon 10), RP2 , RPGR , SNRNP200 |
| 23 | 9 | Retinitis pigmentosa | 6 years | Yes – AD | N/A |
| 24 | 54 | Stargardt disease | Mid-30s | Yes – AR | N/A |
| 25 | 11 | Retinitis pigmentosa | 9 years | Yes – XL | RPGR c.2323_2324delGA (p.Arg775fs) |
| 26 | 44 | Retinitis pigmentosa | ∼42 years | No | N/A |
Fifteen of 26 participants (∼58%) had clearly deleterious mutations or highly suspicious variants of unknown significance consistent with their phenotype. A complete summary of previous genetic testing and exome sequencing results for each participant is shown in Table 2 . Each of these variants was confirmed using Sanger sequencing. Participant 4 had 2 different medically actionable incidental findings: an apparently novel nonsense variant in BRCA2 , c.2857G>T (p.Glu593*), which is associated with hereditary breast ovarian cancer syndrome; and an apparently novel frameshift variant in MSH6 , c.1170delT (p.Phe391fs), which is associated with Lynch syndrome. Both truncating variants were considered likely pathogenic, given their suspected effect on the protein. When questioned further about family history of cancer at results disclosure, he reported that his paternal grandfather had a history of colon cancer at an older age ( Figure ).
| Participant | Exome sequencing Diagnostic Results | Previous Reports | Minor Allele Frequency a | Conservation | Condel Score b | Interpretation | |
|---|---|---|---|---|---|---|---|
| 1 | NC_000003.11:g.129252554C>T | RHO c.1040C>T (p.Pro347Leu) | Pathogenic | 8.263e-06 | Highly conserved | 0.698 Deleterious | Known pathogenic |
| 2 | Negative | ||||||
| 3 | NC_000019.9:g.48343048G>A | CRX c.724G>A (p.Val242Met) c | Pathogenic | 0.001435 | Not conserved | 0.853 Deleterious | Likely benign |
| 4 | NC_000001.10:g.94528806A>G | ABCA4 c.1622T>C (p.Leu541Pro) | Pathogenic | 0.0001235 | Highly conserved | 1.000 Deleterious | Known pathogenic |
| NC_000001.10:g.94528265C>T | ABCA4 c.1805G>A (p.Arg602Gln) | Pathogenic | 1.68e-05 | Highly conserved | 0.451 Deleterious | Known pathogenic | |
| NC_000001.10:g.94508969G>A | ABCA4 c.3113C>T (p.Ala1038Val) | Polymorphic Pathogenic | 0.001426 | Mostly conserved | 0.000 Neutral | Uncertain variant | |
| NC_000013.10:g.32911349G>T | BRCA2 c.2857G>T (p.Glu593*) | None | 0 | — | — | Likely pathogenic | |
| NC_000002.11:g.48026292_48026292delT | MSH6 c.1170delT (p.Phe391fs) | None | 0 | — | — | Likely pathogenic | |
| 5 | Negative | ||||||
| 6 | Negative | ||||||
| 7 | NC_000001.10:g.94466624C>T | ABCA4 c.6320G>A (p.Arg2107His) | Pathogenic | 0.001872 | Highly conserved | 1.000 Deleterious | Known pathogenic |
| NC_000001.10:g.94496666G>A | ABCA4 c.4139C>T (p.Pro1380Leu) | Pathogenic | 0.0002012 | Highly conserved | 0.619 Deleterious | Known pathogenic | |
| NC_000001.10:g.94520708A>G | ABCA4 c.2546T>C (p.Val849Ala) | Pathogenic | 0.001335 | Not conserved | 0.007 Neutral | Uncertain variant | |
| 8 | Negative | ||||||
| 9 | NC_000007.13:g.33380565C>G | BBS9 c.1255C>G (p.Pro419Ala) | None | 9.078e-05 | Mostly conserved | 0.026 Neutral | Uncertain variant |
| NC_000007.13:g.33545217A>T | BBS9 c.2138A>T (p.Glu713Val) | None | 0 | Mostly conserved | 0.922 Deleterious | Uncertain variant | |
| 10 | NC_000001.10:g.94577135C>T | ABCA4 c.161G>A (p.Cys54Tyr) | Pathogenic | 1.65e-05 | Mostly conserved | 0.026 Neutral | Uncertain variant |
| NC_000004.11:g.16008270C>T | PROM1 c.1345G>A (p.Val449Met) | None | 0.001475 | Mostly conserved | 0.810 Deleterious | Likely benign d | |
| 11 | Negative | ||||||
| 12 | NC_000001.10:g.216108128T>C | USH2A c.7130A>G (p.Asn2377Ser) | VUS Polymorphism | 0.003772 | Mostly conserved | 0.001 Neutral | Uncertain variant |
| NC_000001.10:g.215901511G>A | USH2A c.11927C>T (p.Thr3976Met) | VUS | 0.0005192 | Highly conserved | 0.966 Deleterious | Uncertain variant | |
| 13 | Negative | ||||||
| 14 | Negative | ||||||
| 15 | NC_000011.9:g.66293652T>G | BBS1 c.1169T>G (p.Met390Arg) – Homozygous | Pathogenic | 0.001484 | Highly conserved | 0.627 Deleterious | Known pathogenic |
| 16 | NC_000014.8:g.68195946G>C | RDH12 c.697G>C (p.Val233Leu) | Pathogenic | 2.491e-05 | Highly conserved | 0.910 Deleterious | Uncertain variant |
| NC_000014.8:g.68200483T>G | RDH12 c.869T>G (p.Val290Gly) | None | 8.246e-05 | Highly conserved | 0.930 Deleterious | Uncertain variant | |
| 17 | Negative | ||||||
| 18 | NC_000001.10:g.216498866_216498867insTGGC | USH2A c.923_924insGCCa (p.His308fs) | Pathogenic | 0.0001074 | — | — | Known pathogenic |
| NC_000001.10:g.215853632_215853633insAA | USH2A c.12152_12153insTT (p.Glu4051fs) | None | 0 | — | — | Likely pathogenic | |
| 19 | NC_000001.10:g.216420460C>A | USH2A c.2276G>T (p.Cys759Phe) | Pathogenic | 0.000784 | Highly conserved | 1.00 Deleterious | Known pathogenic |
| NC_000001.10:g.215848961A>T | USH2A c.12295-3T>A | None | — | — | Uncertain variant | ||
| 20 | NC_000008.10:g.55538471C>T | RP1 c.2029C>T (p.R677X) | Pathogenic | 0 | — | — | Known pathogenic |
| 21 | Negative | ||||||
| 22 | NC_000007.13:g.128034615C>T | MERTK c.1787-2A>C Apparently homozygous e | None | 0 | — | — | Likely pathogenic |
| 23 | Negative | ||||||
| 24 | NC_000006.11:g.42689616T>C | PRPH2 c.457A>G (p.Lys153Glu) | None | 0 | Highly conserved | 0.873 Deleterious | Uncertain variant |
| 25 | NC_000023.10:g.38145928_38145929delCT | RPGR c.2323_2324delGA (p.Arg775fs) | Pathogenic | 0 | — | — | Known pathogenic |
| 26 | NC_000001.10:g.94528143C>T | ABCA4 c.1927G>A (p.Val643Met) | Pathogenic | 0.001618 | Highly conserved | 0.383 Neutral | Uncertain variant |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


