Hereditary Optic Neuropathies
The hereditary optic neuropathies comprise a group of disorders in which the cause of optic nerve dysfunction is hereditable as demonstrated by familial expression or genetic analysis. Clinical variability, both within and among families with the same disease, often makes recognition and classification difficult. Previously, classification relied on recognition of similar characteristics and similar patterns of transmission, but genetic analysis now permits diagnosis of the hereditary optic neuropathies in the absence of family history or in the setting of unusual clinical presentations. As a result, the clinical phenotypes of each disease are broader, and it is easier to recognize unusual cases.
The inherited optic neuropathies typically manifest as symmetric, bilateral, and central visual loss. In many of these disorders, the papillomacular nerve fiber bundle is exclusively or primarily affected, with resultant central or cecocentral scotomas. The exact location of initial pathology along the ganglion cell and its axon, and the pathophysiologic mechanisms of optic nerve injury remain unknown. Optic nerve damage usually is permanent and, in many diseases, progressive. Once optic atrophy is observed, substantial nerve injury has already occurred although in rare settings, at least partial recovery can still occur.
In classifying the hereditary optic neuropathies, it is important to exclude the primary retinal degenerations that may masquerade as primary optic neuropathies because of the common finding of secondary optic disc pallor (see Chapter 2).
The inherited optic neuropathies generally are classified by their pattern of transmission. The most common patterns are autosomal dominant, autosomal recessive, and maternal (i.e., mitochondrial). In some of the hereditary optic neuropathies, optic nerve dysfunction is, or at least usually is the only manifestation of the disease. In others, a bilateral optic neuropathy is associated with various, apparently disparate neurologic deficits and/or systemic abnormalities. In some of these disorders, both the visual loss and the nonvisual deficits are static, whereas in others, either or both are progressive. Finally, in some patients, a hereditary optic neuropathy is simply a consequence of the optic nerve being a white-matter tract in a patient with a diffuse white-matter disorder or degeneration.
Monosymptomatic Hereditary Optic Neuropathies
Leber Hereditary Optic Neuropathy
Clinical Features
In this maternally inherited disorder (see below), men become symptomatic more frequently than women, with a male predominance of 80% to 90% in most pedigrees. There is incomplete penetrance, with 20% to 60% of men and 4% to 32% of women with one of the causative genetic mutations experiencing visual loss. Affected females are more likely to have affected children, especially daughters, than unaffected female carriers.
Visual loss in patients with Leber hereditary optic neuropathy (LHON) typically begins simultaneously in both eyes (50%) or initially in one eye, followed days, weeks, months or, very rarely, years after the first eye (50%). Reports of unilateral onset are numerous and likely reflect both instances of true unilateral visual loss and cases in which initial involvement of the contralateral eye is unrecognized by the patient.
Indeed, careful visual field testing in apparently unilateral involvement usually shows a subtle field defect in the contralateral asymptomatic eye. In any event, all patients with LHON eventually experience bilateral and symmetric visual loss. Once visual loss begins, it tends to worsen, reaching a nadir in 3 to 6 months. Visual acuity at the point of stabilization usually is about 20/200 to 20/400. Although vision in some patients deteriorates to hand motions, it is distinctly unusual for a patient to have a reduction to light perception or no light perception, and in such cases, the diagnosis of LHON must be questioned, as it must be in patients whose visual loss is minimal (e.g., 20/40 OU).
Indeed, careful visual field testing in apparently unilateral involvement usually shows a subtle field defect in the contralateral asymptomatic eye. In any event, all patients with LHON eventually experience bilateral and symmetric visual loss. Once visual loss begins, it tends to worsen, reaching a nadir in 3 to 6 months. Visual acuity at the point of stabilization usually is about 20/200 to 20/400. Although vision in some patients deteriorates to hand motions, it is distinctly unusual for a patient to have a reduction to light perception or no light perception, and in such cases, the diagnosis of LHON must be questioned, as it must be in patients whose visual loss is minimal (e.g., 20/40 OU).
The visual loss in patients with LHON is always painless, and it is the absence of pain that is a major feature that differentiates LHON from other optic neuropathies, particularly optic neuritis, which occurs in the same general age group (although more often in women than in men, the opposite of LHON) but is associated with pain, often on movement of the eye, in over 90% of cases.
In addition to reduction of visual acuity, color vision is severely affected in patients with LHON, often early in the course but rarely if ever before there is significant loss of acuity. Pupillary light responses may be relatively preserved compared with the responses in patients with optic neuropathies from other causes. Indeed, even patients with apparently unilateral visual loss from LHON tend to have either no relative afferent pupillary defect or a very subtle one. This is another feature that differentiates LHON from other optic neuropathies and is thought to be related to selective sparing of the melanopsin-containing retinal ganglion cells in this disorder (see Chapter 2).
Visual field defects in patients with LHON typically are central or cecocentral. The scotomas may be relative during the early stages of visual loss but rapidly become large and absolute. Rarely, field abnormalities mimicking the bitemporal configuration of chiasmal defects occur.
Many patients with LHON have absolutely normal-appearing optic discs and peripapillary retinas at the time visual loss begins and, thus, may be thought to have either retinal disease or nonorganic visual loss, particularly given their relatively preserved pupillary responses to light stimulation and lack of pain. Others, however, have the triad of: (1) optic disc and circumpapillary telangiectasias, (2) hyperemia and fullness of the optic disc (or discs if visual loss is bilateral) without peripapillary hemorrhages or exudates, and (3) absence of leakage from the disc on fluorescein angiography (distinguishing the LHON optic disc appearance from the truly swollen optic disc) (Fig. 11.1). When present, these findings at the time of visual loss are helpful in suggesting the correct diagnosis, but it must be emphasized that their absence—even during the period of acute visual loss—does not exclude the diagnosis of LHON. As visual loss progresses in patients with LHON, the optic disc becomes pale, particularly temporally, with loss of nerve fibers that is most pronounced in the papillomacular region, producing a typical picture similar to that of patients with severe toxic or nutritional optic neuropathies (Fig. 11.2; see also Chapter 10).
Visual loss in most patients with LHON remains profound and permanent; however, recovery of central vision may occasionally occur after visual deterioration. Unlike the spontaneous improvement in vision that occurs in patients with optic neuritis within several months after onset, spontaneous visual improvement in patients with LHON (4–30%, depending on the mutation–see below) usually occurs about 1 to 2 years later, typically associated with a partial breakup or, rarely, complete resolution of the central field defects. Recovery usually is bilateral and symmetric, and once it occurs, recurrent visual loss is extremely rare.
Associated Findings
In the majority of patients with LHON, visual dysfunction is the only significant manifestation of the disease; however, some pedigrees have members with associated cardiac conduction abnormalities, such as the pre-excitation syndromes (specifically Wolff–Parkinson–White and Lown–Ganong–Levine) and prolongation of the corrected QT interval. Rarely, such patients experience palpitations, syncope, or sudden death. Minor neurologic abnormalities, such as exaggerated or pathologic reflexes, mild cerebellar ataxia, tremor, movement disorders, muscle wasting, or distal sensory neuropathy, are present in some patients with LHON. In addition, some patients with molecularly confirmed LHON, predominantly women, exhibit symptoms and signs consistent with multiple sclerosis (MS) at the time they begin to experience the typical painless, progressive visual loss typical of LHON. Cerebrospinal fluid (CSF) and magnetic resonance imaging (MRI) findings are characteristic of MS in most if not all of these patients. It therefore is likely that this apparent association of LHON and MS is no greater than the prevalence of the two diseases.
Some pedigrees consist of multiple members who exhibit the clinical features of LHON in addition to more severe neurologic abnormalities, the so-called “Leber plus” syndromes. In addition to the typical bilateral optic neuropathy, these syndromes include: (1) movement disorders, spasticity, psychiatric disturbances, skeletal abnormalities, and acute infantile encephalopathy; (2) dystonia, and lesions of the basal ganglia by neuroimaging; optic neuropathy and myelopathy; and (3) fatal encephalopathy in early childhood. Most of these pedigrees are genetically
distinct from those with more classic LHON, with some having more than one of the mutations typically associated with this disorder.
distinct from those with more classic LHON, with some having more than one of the mutations typically associated with this disorder.
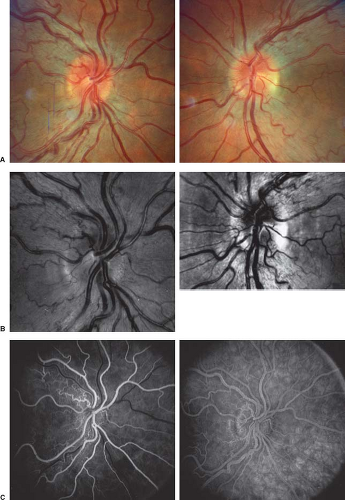 Figure 11.1 Leber hereditary optic neuropathy. A: Both optic discs in the acute phase of the disorder. Note hyperemic appearance of discs. Peripapillary telangiectatic vessels are present. B: Both optic discs photographed with red-free (540 nm) light. Note marked hyperemia of the discs with dilation of small vessels both on the surface of the disc and in the peripapillary region. C: Fluorescein angiogram in arteriovenous (left) and late (right) phases shows dilation of right disc and peripapillary vessels but no leakage of fluorescein dye. |
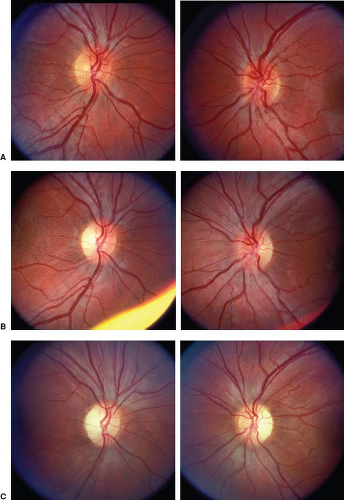 Figure 11.2 Progression to bilateral optic atrophy in a patient with Leber hereditary optic neuropathy. A: Both optic discs in acute phase of the disease. Visual acuity is 20/100 in both eyes with large cecocentral scotomas. Telangiectatic vessels are evident adjacent to the inferior margin of both discs. The right disc (left) is already pale temporally, and there is atrophy of the papillomacular nerve fiber layer. B: Two months after onset of visual loss, visual acuity is 5/200 in the right eye and 8/200 in the left eye. Both discs show moderate pallor, particularly temporally, with loss of nerve fiber layer that is especially evident in the papillomacular region. Previously seen telangiectatic vessels are disappearing. C: Six months after onset of visual loss, visual acuity remains 5/200 in the right eye and 8/200 in the left eye. Both discs are pale, particularly temporally. The telangiectatic vessels have completely disappeared. One year after these photographs were obtained, the patient noted gradual, partial return of visual acuity in both eyes to 20/50. Subsequent genetic testing showed evidence of a mutation in the mitochondrial DNA at position 14484. |
Inheritance and Genetics
All pedigrees clinically designated as LHON have a maternal inheritance pattern; that is, all offspring of a woman carrying the trait will inherit the trait, but only the females can pass the trait on to the subsequent generation. Both the father and the mother contribute to the nuclear portion of the zygote, but the mother’s egg is essentially the sole provider of the cytoplasmic contents of the zygote. Therefore, a cytoplasmic determinant is necessary for maternal inheritance, and the only source of extranuclear DNA in the cell is in the intracytoplasmic mitochondria.
Almost every cell in the body contains several hundred intracytoplasmic mitochondria that generate the cellular energy necessary for normal cellular function and maintenance. Those cells in tissues particularly reliant on mitochondrial energy production, such as the central nervous system (CNS), contain more mitochondria than cells with low energy requirements. Each mitochondrion contains 2 to 10 double-stranded circles of DNA, with each circle containing 16,500 base pairs compared with the 3 × 109 base pairs contained within the nuclear genome. Because there are several mtDNAs in each mitochondrion and hundreds of mitochondria per cell, the mtDNA comprises approximately 0.3% of the cell’s total DNA.
Mitochondrial DNA codes for all the transfer ribonucleic acids (RNAs) and the ribosomal RNAs required for intramitochondrial protein production. It also codes for 13 proteins essential to the oxidative phosphorylation system. The majority of proteins crucial to normal oxidative phosphorylation function are encoded on nuclear genes, manufactured in the cytoplasm, and transported into the mitochondria. Hence, “mitochondrial disease” can result from genetic defects in either the nuclear or mitochondrial genomes.
If a new mutation occurs in the mtDNA, there will be a period of coexistence of mutant and normal mtDNA within the same cell (heteroplasmy). At each cell division, the mitochondrial genotype may drift toward pure normal or pure mutant (homoplasmy), or it may remain mixed. The phenotype of the cell (and the tissue the cells comprise) depends on the proportion of mtDNA genotypes and the intrinsic energy needs of the cell. The mutant phenotype may only become apparent when the amount of normal mtDNA can no longer provide sufficient mitochondrial function for cell and tissue maintenance.
The most common point mutation in mtDNA linked to LHON is a single nucleotide substitution at position 11778 in the mtDNA (Fig. 11.3). This region codes for subunit 4 (designated ND4) of complex I (NADH dehydrogenase) in the respiratory chain. The 11778 mutation is present in many racially divergent pedigrees with LHON, suggesting that it has arisen independently on multiple occasions. Whereas 31% to 89% of European, North American, and Australian LHON pedigrees have the 11778 mutation, greater than 90% of Asian LHON patients are 11778 positive.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


