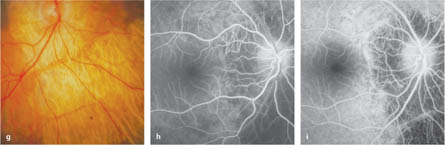
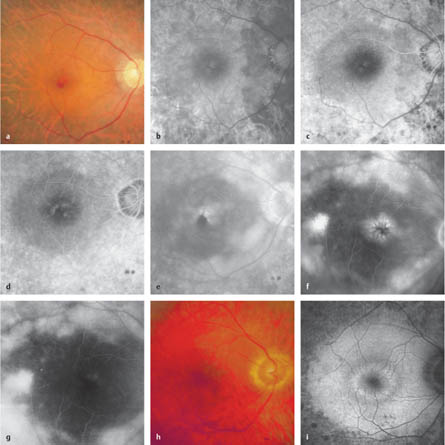
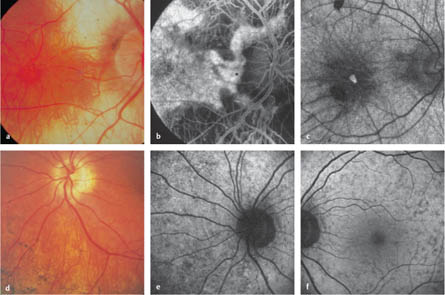
4.1 Retinitis Pigmentosa Gass JD. Retinitis pigmentosa (rod-cone dystrophies). In: Gass JD, ed. Stereoscopic atlas of macular diseases: diagnosis and treatment, 4th ed. St. Louis: Mosby, 1997: 352–8. Rivolta C, Sharon D, DeAngelis MM, Dryja TP. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet 2002;11:1219–27. Robson AG, Egan CA, Luong VA, et al. Comparison of fundus autofluorescence with photopic and scotopic fine-matrix mapping in patients with retinitis pigmentosa and normal visual acuity. Invest Ophthalmol Vis Sci 2004;45:4119–25 Weleber RG, Gregory-Evans K. Retinitis pigmentosa and allied disorders. In: Ryan SJ, ed. Retina, 4th ed. Philadelphia: Elsevier, 2006:395–498. Fig. 4.1a–i Retinitis pigmentosa a Color photograph. Constricted vessels, peripheral choriocapillaris atrophy, and a cystoid macular edema that almost has the appearance of a macular hole. b Early phase. Large choroidal vessels are visible in the area of the choriocapillaris atrophy. c Late early phase. An oval area with a relatively conserved retinal pigment epithelium can be seen at the posterior pole. d Early late phase. Initial perifoveal leakage. e Late phase. Cystoid macular edema and choroidal hyperfluorescence in the peripheral areas, with choriocapillary and retinal pigment epithelial loss. f Late phase (in a different patient). Cystoid macular edema. g Late phase (in the same patient as in f). Reduction in the cystoid macular edema after oral acetazolamide therapy. h Color photograph. Unobtrusive pigment epithelium irregularities at the posterior pole. i Fundus autofluorescence. Peripheral destruction of the retinal pigment epithelium and a pericentral ring of increased autofluorescence. Do DV, Zhang K, Garabaldi DC, Carr RE, Sunness JS. Hereditary choroidal disease. In: Ryan SJ, ed. Retina, 4th ed. Philadelphia, Elsevier, 2006:499–508. Gass JD. Atypical forms of retinitis pigmentosa. In: Gass JD, ed. Stereoscopic atlas of macular diseases: diagnosis and treatment, 4th ed. St. Louis: Mosby, 1997:358–81. Kaiser-Kupfer MI, Caruso RC, Valle D, Reed GF. Use of an arginine-restricted diet to slow progression of visual loss in patients with gyrate atrophy. Arch Ophthalmol 2004;122:982–4. Roberts MF, Fishman GA, Roberts DK, et al. Retrospective, longitudinal, and cross sectional study of visual acuity impairment in choroideraemia. Br J Ophthalmol 2002;86:658–62. Fig. 4.2a–f Choroideremia a Color photograph. Temporal and subfoveal to the disk, a patch of choriocapillaris is still present; more peripherally, only a few large choroidal vessels can be detected. b Arteriovenous phase. Only a few large choroidal vessels are visible in the atrophic areas; retained choriocapillaris and pigment epithelium with pigment epithelium defects can be seen at the posterior pole and temporal to the optic disc. c Fundus autofluorescence (in a different patient). A small island of retained pigment epithelium is detectable through a patch of remaining autofluorescence. d Color photograph in a female carrier. Several variably distinct pigmentations can be seen, while visual acuity, color vision, the field of vision, and electroretinography were normal. e Fundus autofluorescence. A speckled pattern with small areas of reduced or increased autofluorescence is visible nasally. f Fundus autofluorescence. At the posterior pole, there is a speckled pattern with small areas of reduced autofluorescence. Fig. 4.2g–i Gyrate atrophy g Color photograph. Sharply delineated peripapillary choroid atrophy, which continues toward the periphery below the optic disc. h Early phase. Only the large choroidal vessels are visible in the affected areas. i Arteriovenous phase. A small bridge of retained choriocapillaris is recognizable below the optic disc. The choroid outside the affected areas appears to be intact.
Epidemiology, Pathophysiology, and Clinical Presentation
 Retinitis pigmentosa is the most common group of retinal dystrophies (with a prevalence of approximately one in 5000).
Retinitis pigmentosa is the most common group of retinal dystrophies (with a prevalence of approximately one in 5000).
 Mutations in at least 32 genes have been associated with the development of nonsyndromic retinitis pigmentosa. It can be expected that additional gene associations will be discovered.
Mutations in at least 32 genes have been associated with the development of nonsyndromic retinitis pigmentosa. It can be expected that additional gene associations will be discovered.
 The products of these genes are either expressed in the retinal pigment epithelium (RPE) or the photoreceptors. Within the photoreceptor–retinal pigment epithelium complex, the degeneration of one cell type is followed by the degeneration of the others. It is not possible to recognize clinically in this process whether the primary damage is located in the photoreceptor or in the RPE. It is usually the rods that are mainly affected, but degeneration of the rods subsequently leads to degeneration of the cones.
The products of these genes are either expressed in the retinal pigment epithelium (RPE) or the photoreceptors. Within the photoreceptor–retinal pigment epithelium complex, the degeneration of one cell type is followed by the degeneration of the others. It is not possible to recognize clinically in this process whether the primary damage is located in the photoreceptor or in the RPE. It is usually the rods that are mainly affected, but degeneration of the rods subsequently leads to degeneration of the cones.
 All patterns of inheritance can occur (autosomal-dominant, autosomal-recessive, X-linked, mitochondrial). In some syndromes, combinations with disorders in other organs are not uncommon (e. g., with hearing impairment in Usher’s syndrome).
All patterns of inheritance can occur (autosomal-dominant, autosomal-recessive, X-linked, mitochondrial). In some syndromes, combinations with disorders in other organs are not uncommon (e. g., with hearing impairment in Usher’s syndrome).
 The time of onset ranges from congenital conditions (as in Leber congenital amaurosis) to courses with very minor abnormalities that may only be identified accidentally in later life. Due to this variability, it is very difficult to assess the prognosis in individual cases.
The time of onset ranges from congenital conditions (as in Leber congenital amaurosis) to courses with very minor abnormalities that may only be identified accidentally in later life. Due to this variability, it is very difficult to assess the prognosis in individual cases.
 Clinically, the course of the disease is characterized by the onset of impaired night vision. Slowly progressive loss of peripheral visual fields develops, often leaving only an isolated central visual field intact. Electroretinography (ERG) may show severely reduced responses or an absence of detectable responses.
Clinically, the course of the disease is characterized by the onset of impaired night vision. Slowly progressive loss of peripheral visual fields develops, often leaving only an isolated central visual field intact. Electroretinography (ERG) may show severely reduced responses or an absence of detectable responses.
Fluorescein Angiography
 Fluorescein angiography is not necessary for the diagnosis of retinitis pigmentosa.
Fluorescein angiography is not necessary for the diagnosis of retinitis pigmentosa.
 Loss of the choriocapillaris and defects in the RPE are already recognizable in the early phase.
Loss of the choriocapillaris and defects in the RPE are already recognizable in the early phase.
 In the late phase, leakage can be observed when cystoid macular edema or vascular abnormalities similar to those in Coats disease are present.
In the late phase, leakage can be observed when cystoid macular edema or vascular abnormalities similar to those in Coats disease are present.
Fundus Autofluorescence
 A reduction in or absence of fundus autofluorescence indicates areas with loss of photoreceptors and/or of the RPE.
A reduction in or absence of fundus autofluorescence indicates areas with loss of photoreceptors and/or of the RPE.
 A ring of increased perifoveal autofluorescence can be observed in some patients. This corresponds to the border of central retinal function. Concentric progression of the ring can be observed over time.
A ring of increased perifoveal autofluorescence can be observed in some patients. This corresponds to the border of central retinal function. Concentric progression of the ring can be observed over time.
 The fovea can show normal, increased, or reduced autofluorescence.
The fovea can show normal, increased, or reduced autofluorescence.
 Further research is needed in order to define the prognostic value of fundus autofluorescence imaging.
Further research is needed in order to define the prognostic value of fundus autofluorescence imaging.
Diagnosis and Treatment
 The initial diagnosis of retinitis pigmentosa is based on perimetry, ophthalmoscopy in mydriasis, and an ERG.
The initial diagnosis of retinitis pigmentosa is based on perimetry, ophthalmoscopy in mydriasis, and an ERG.
 Even if typical ophthalmoscopic findings are present, at least one ERG recording is recommended, as there may be significant discrepancies between the morphological and functional findings and assessment of retinal function provides the best basis for counseling the patient.
Even if typical ophthalmoscopic findings are present, at least one ERG recording is recommended, as there may be significant discrepancies between the morphological and functional findings and assessment of retinal function provides the best basis for counseling the patient.
 Fundus autofluorescence may detect morphological alterations that are not visible with ophthalmoscopy.
Fundus autofluorescence may detect morphological alterations that are not visible with ophthalmoscopy.
 Fluorescein angiography is only indicated in the presence of abnormal Coats-like vessels or when cystoid macular edema is suspected. The latter can also be diagnosed with optical coherence tomography. This can be particularly recommended, as severe cystoid macular edema may resemble macular hole formation.
Fluorescein angiography is only indicated in the presence of abnormal Coats-like vessels or when cystoid macular edema is suspected. The latter can also be diagnosed with optical coherence tomography. This can be particularly recommended, as severe cystoid macular edema may resemble macular hole formation.
 Cystoid macular edema may respond well to oral acetazolamide administration.
Cystoid macular edema may respond well to oral acetazolamide administration.
 Photocoagulation limits the progression of abnormal vessel formation in the few patients with Coats-like vessels.
Photocoagulation limits the progression of abnormal vessel formation in the few patients with Coats-like vessels.
 There are as yet no means of treating the condition, although clinical trials of gene therapy are being prepared.
There are as yet no means of treating the condition, although clinical trials of gene therapy are being prepared.
 Refractive errors should be corrected and, where applicable, the use of special filter glasses and low-vision aids can be very helpful.
Refractive errors should be corrected and, where applicable, the use of special filter glasses and low-vision aids can be very helpful.
 Nutritional supplements (e. g., vitamin A palmitate, docosahexaenoic acid) may delay the progression of some forms of retinitis pigmentosa.
Nutritional supplements (e. g., vitamin A palmitate, docosahexaenoic acid) may delay the progression of some forms of retinitis pigmentosa.
 Early cataract formation is frequent and requires surgery, as subcapsular cataracts typically develop on the posterior pole of the lens, directly in front of the remainder of the central visual field.
Early cataract formation is frequent and requires surgery, as subcapsular cataracts typically develop on the posterior pole of the lens, directly in front of the remainder of the central visual field.
References
4.2 Choroideremia and Gyrate Atrophy
Choroideremia
Epidemiology, Pathophysiology, and Clinical Presentation
 An X-linked condition, choroideremia is a generalized form of choroidal dystrophy that starts in the mid-periphery of the retina and is associated with mutations in the CHM (REP-1) gene.
An X-linked condition, choroideremia is a generalized form of choroidal dystrophy that starts in the mid-periphery of the retina and is associated with mutations in the CHM (REP-1) gene.
 As in retinitis pigmentosa, symptoms start with night blindness and mid-peripheral visual field loss with progressive concentric narrowing, whereas visual acuity and color vision remain very good for a longer period of time.
As in retinitis pigmentosa, symptoms start with night blindness and mid-peripheral visual field loss with progressive concentric narrowing, whereas visual acuity and color vision remain very good for a longer period of time.
 In males, fine pigment-epithelial irregularities can be observed initially, followed by small mid-peripheral areas of choroidal atrophy with fuzzy edges, which later converge. A central island of intact choroid often persists for a longer period at the posterior pole. The retinal vessels usually change very little.
In males, fine pigment-epithelial irregularities can be observed initially, followed by small mid-peripheral areas of choroidal atrophy with fuzzy edges, which later converge. A central island of intact choroid often persists for a longer period at the posterior pole. The retinal vessels usually change very little.
 In female carriers, distinct pigment changes are almost always present over the whole retina. Retinal function is generally normal. Symptomatic female carriers with a disease progression similar to that in males are rare.
In female carriers, distinct pigment changes are almost always present over the whole retina. Retinal function is generally normal. Symptomatic female carriers with a disease progression similar to that in males are rare.
Fluorescein Angiography
 Fluorescein angiography allows early detection of affected choroidal areas.
Fluorescein angiography allows early detection of affected choroidal areas.
 As in the clinical findings, choriocapillaris atrophy with fuzzy edges can be seen with angiography. The borders of atrophic areas are lined with increased fluorescence in the late phase.
As in the clinical findings, choriocapillaris atrophy with fuzzy edges can be seen with angiography. The borders of atrophic areas are lined with increased fluorescence in the late phase.
Fundus Autofluorescence
 Fundus autofluorescence can identify areas of retinal pigment epithelial loss in males.
Fundus autofluorescence can identify areas of retinal pigment epithelial loss in males.
 In females, fundus autofluorescence shows a speckled pattern, with small areas of reduced autofluorescence. This autofluorescence pattern may suggest carrier status as an incidental finding in women who do not have a family history of the condition.
In females, fundus autofluorescence shows a speckled pattern, with small areas of reduced autofluorescence. This autofluorescence pattern may suggest carrier status as an incidental finding in women who do not have a family history of the condition.
Diagnosis and Treatment
 In men with the early stages of choroideremia, with alterations in the retinal pigment epithelium, it is possible for the condition to be confused with retinitis pigmentosa.
In men with the early stages of choroideremia, with alterations in the retinal pigment epithelium, it is possible for the condition to be confused with retinitis pigmentosa.
 More serious, however, and more burdensome for the patients affected, is misdiagnosis of this condition as retinitis pigmentosa, on the basis of the changes in the retina, in female carriers—since these patients do not generally experience any relevant functional disturbances. Fundus autofluorescence is recommended for differential diagnosis.
More serious, however, and more burdensome for the patients affected, is misdiagnosis of this condition as retinitis pigmentosa, on the basis of the changes in the retina, in female carriers—since these patients do not generally experience any relevant functional disturbances. Fundus autofluorescence is recommended for differential diagnosis.
 Full-field electroretinography (ERG) shows that in men, rod-dependent responses are more reduced than cone-dependent ones. In women, the ERG is generally normal. In contrast, the fundus is usually normal in female carriers of X-linked retinitis pigmentosa, but the amplitudes of the ERG are more often reduced in these cases.
Full-field electroretinography (ERG) shows that in men, rod-dependent responses are more reduced than cone-dependent ones. In women, the ERG is generally normal. In contrast, the fundus is usually normal in female carriers of X-linked retinitis pigmentosa, but the amplitudes of the ERG are more often reduced in these cases.
 Molecular-genetic diagnosis is possible, but this is complex and is not necessary in clear-cut cases.
Molecular-genetic diagnosis is possible, but this is complex and is not necessary in clear-cut cases.
 There are no causal treatments.
There are no causal treatments.
 It is important to exclude gyrate atrophy, a less frequent condition, during differential diagnosis.
It is important to exclude gyrate atrophy, a less frequent condition, during differential diagnosis.
Gyrate Atrophy
Epidemiology, Pathophysiology, and Clinical Presentation
 Gyrate atrophy is an autosomal-recessive condition involving generalized choroidal dystrophy that starts in the mid-periphery of the retina and is associated with mutations in the ornithine aminotransferase gene (OAT).
Gyrate atrophy is an autosomal-recessive condition involving generalized choroidal dystrophy that starts in the mid-periphery of the retina and is associated with mutations in the ornithine aminotransferase gene (OAT).
 In contrast to choroideremia, the borders of the atrophic areas are sharply delineated.
In contrast to choroideremia, the borders of the atrophic areas are sharply delineated.
 Otherwise, the clinical course is similar to that in choroideremia, with confluence of patchy choroidal atrophy and progression to the posterior pole. In addition, early cataract formation is observed.
Otherwise, the clinical course is similar to that in choroideremia, with confluence of patchy choroidal atrophy and progression to the posterior pole. In addition, early cataract formation is observed.
Fluorescein Angiography
 Fluorescein angiography reveals choriocapillaris atrophy, with sharply delineated edges and increased fluorescence on the border of the lesions.
Fluorescein angiography reveals choriocapillaris atrophy, with sharply delineated edges and increased fluorescence on the border of the lesions.
Diagnosis and Treatment
 The ERG shows that rod-dependent responses are more reduced than cone-dependent ones.
The ERG shows that rod-dependent responses are more reduced than cone-dependent ones.
 Molecular-genetic diagnosis is possible, but is not necessary in clear-cut cases.
Molecular-genetic diagnosis is possible, but is not necessary in clear-cut cases.
 Markedly elevated ornithine blood levels are pathognomonic.
Markedly elevated ornithine blood levels are pathognomonic.
 A specific diet may influence the course of the disease.
A specific diet may influence the course of the disease.
References
4.3 X-Linked Congenital Retinoschisis
Epidemiology, Pathophysiology, and Clinical Presentation
 X-linked retinoschisis is a frequent form of retinal dystrophy in young males associated with mutations in the RS1 gene. The RS1 gene product is important for cell-to-cell interaction in the retina.
X-linked retinoschisis is a frequent form of retinal dystrophy in young males associated with mutations in the RS1 gene. The RS1 gene product is important for cell-to-cell interaction in the retina.
 The morphological changes vary from severe retinoschisis affecting the complete retina at 3 months of age to unobtrusive foveal alterations that are not infrequently overlooked during ophthalmoscopic examinations.
The morphological changes vary from severe retinoschisis affecting the complete retina at 3 months of age to unobtrusive foveal alterations that are not infrequently overlooked during ophthalmoscopic examinations.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


