Chronic rhinosinusitis (CRS) is a prevalent condition that is heterogeneous in disease characteristics and multifactorial in cause. Although sinonasal mucosal inflammation in CRS is often either reversible or well-managed medically and surgically, a significant proportion of patients has a refractory form of CRS despite maximal therapy. Two of the several described factors thought to contribute to disease recalcitrance are genetic influences and dysfunction of the host immune system. Current evidence for a genetic basis of CRS is reviewed, as it pertains to putative abnormalities in innate and adaptive immune function. The role of systemic immunodeficiencies in refractory CRS is discussed.
Key points
- •
Local defects in innate and adaptive immunity have been implicated in the pathogenesis of refractory chronic rhinosinusitis (CRS).
- •
A genetic basis in CRS is suggested by an association with well-defined heritable disorders as well as limited evidence of a familial inheritance pattern.
- •
More than 445 single-nucleotide polymorphisms have been associated with CRS, although these largely have not been replicated.
- •
Systemic immune dysregulation in refractory CRS may be demonstrated with laboratory testing of humoral and cell-mediated immunity.
Introduction
Chronic rhinosinusitis (CRS) is defined in adults as persistent symptoms of sinonasal inflammation for 12 or more weeks with confirmatory objective findings on nasal endoscopy or computed tomography (CT). CRS is a multifactorial disease with proposed etiopathologies including occupational/environmental exposures, infection, immune dysfunction, and genetic predisposition. As understanding of the disease evolves, clinical phenotypes have emerged that allow subclassification within the entity once identified solely as CRS. The presence or absence of polyps is used as a primary distinction because of easily recognizable clinical features and underlying inflammatory profiles. CRS with nasal polyps (CRSwNP) is characterized by T helper (Th)-2 polarization, with eosinophilia, increased levels of interleukin-4 (IL-4), IL-5, and IL-13, as well as high local production of immunoglobulin E (IgE). CRS without nasal polyps (CRSsNP) demonstrates a mixed cytokine profile that lacks Th2 polarization. High levels of interferon-γ (IFN-γ) and transforming growth factor-β have been reported in CRSsNP, but the consistency of this feature continues to be under investigation. Although Th2 polarization clearly characterizes CRSwNP in Western countries, evidence suggests that Th2 skewing may be absent in CRSwNP patients in China. This difference may be disappearing as the region undergoes modernization. A third distinct phenotype of CRS is aspirin-exacerbated respiratory disease (AERD), which is fully discussed elsewhere in this issue.
Clinically, otolaryngologists encounter refractory forms of rhinosinusitis within each phenotype. No consensus statement defining recalcitrant CRS exists; however, broadly speaking, CRS is considered refractory when it responds poorly to medical and surgical therapy. Multiple theories and possible pathophysiologic mechanisms have been proposed to explain recalcitrance in CRS. In this review, the authors focus specifically on the contribution of genetics and immunologic dysregulation.
At the most basic level, support for an underlying genetic contribution to CRS stems from observations of familial inheritance patterns. Most of these observations were made in patients with nasal polyps, and, therefore, conclusions about the familial inheritance pattern of CRSsNP cannot be drawn. In patients with nasal polyps, a heritability of 14% to 42% has been described. Cohen and colleagues subclassified CRSwNP patients into 3 groups: isolated polyps, polyps with asthma, and AERD. Interestingly, they found that AERD had the highest heritability (42%) followed by asthma and polyps (30%) and isolated polyps (15%). In addition, they demonstrated a correlation with the number of family members (frequency) with nasal polyposis and the severity of disease showing that severity is proportional to the penetrance of an underlying genetic component.
As a caveat, growing evidence suggests that multiple endotypes of CRS, yet to be fully elucidated, may be present beyond the CRSsNP/CRSwNP/AERD classification scheme. Recognition that CRS remains a complex and incompletely defined disease has important consequences for weighing the validity of genetic and immunologic studies. Without precise definitions of CRS subtypes, meaningful interpretation of research findings is difficult, particularly when detailed clinical information, including raw laboratory and radiographic findings, is not available. At present, differences among CRS subtypes are likely obscured by classifications based on an incomplete understanding, and current scoring systems used for clinical research do not capture CRS features with sufficient granularity. Because of the variety within CRS, more meticulous gathering of patient data with subclassification will almost certainly be necessary to detect true genetic and immunologic contributors. Even if precise categorization were possible, CRS fluctuates in severity and characteristics over time and can develop later in life, so flawed subtype assignments are possible, particularly in genetic studies. All of these challenges must be appreciated, and the existing CRS literature should be evaluated through this lens.
Innate Immunity and Epithelial Barrier Dysfunction in Chronic Rhinosinusitis
Among the mechanisms proposed to contribute to CRS pathogenesis and recalcitrance, many involve abnormalities of local mucosal immune defense. Sinonasal innate immune function begins with the physical epithelial barrier, mucus production, and mucociliary clearance, extending to involve secreted antimicrobial factors and phagocytic hematopoietic cells that combat infection in the airway lumen. Multiple inflammatory mediators produced by epithelial cells and other nonlymphocytic cell populations interact with the adaptive immune system to drive antigen-specific antibody production. Although lymphocytes had long been assumed to be the principal drivers of chronic inflammation, it has become increasingly apparent that tissue-bound epithelial cells, fibroblasts, macrophages, dendritic cells, mast cells, and innate lymphoid cells (ILCs) also participate in perpetuating inflammation. These cell types lack the ability to generate antigen-specific receptors through gene recombination, but instead use germline-encoded pattern recognition receptors (PRR) that bind to microbial elements and to damage-associated molecules. Genetic aberration or dysregulation of any of these innate immune pathways has the potential to contribute to CRS recalcitrance, and there is a growing body of evidence to support this concept. Abnormal mucus viscoelasticity and diminished mucociliary clearance are linked to CRS, including in genetically based diseases such as cystic fibrosis (CF) and primary ciliary dyskinesia. Alteration in expression of PRRs such as toll-like receptors (TLRs) and taste receptors may also be associated with refractory CRS.
Several antimicrobial products are present in mucus and the sinonasal mucosa and include:
- •
Acute phase proteins
- •
Neutralizing proteins
- •
Enzymes
- •
Opsonins
- •
Defensins
- •
Protease inhibitors
- •
Surfactants
Increased levels of surfactant protein A, lysozyme, C3, and C5 have been demonstrated in tissue specimens of CRSsNP compared with controls. In CRSwNP, increased levels of lysozyme, C3, cathelicidin, and acidic mammalian chitinase have been demonstrated compared with controls. Other studies have shown decreased levels of antimicrobial products in both CRSsNP and CRSwNP. Altogether, these findings indicate that abnormal functioning or regulation of the innate immune system is present in both CRSsNP and CRSwNP and may play a significant role in the persistent inflammation that is a hallmark of all CRS.
PRRs may be activated by pathogen-associated molecular patterns or damage-associated molecular patterns. These receptors are found on most cell types, including macrophages, lymphocytes, and epithelial cells. Interestingly, studies have shown that TLR2, TLR4, and TLR7 messenger RNA (mRNA) and protein levels are significantly lower in CRSsNP than controls. Similar findings of altered expression of TLR2, TLR4, TLR7, and TLR9 have been demonstrated in sinonasal tissue from patients with CRSwNP. Another class of PRRs, taste receptors, has been shown to be expressed by nasal solitary chemosensory cells and to modulate epithelial cell innate immune activity in vitro. Dysregulation of taste receptors in CRS has been suggested but not demonstrated. Although some studies have shown downregulation of PRRs and others have shown upregulation of the same PRRs, these data in aggregate imply that dysfunction or dysregulation of innate immunity is present and potentially contributing to the pathogenesis of CRS.
One example of potential innate immune dysregulation in refractory CRS is decreased expression and function of the PRR TLR9. TLR9 activation leads to the production and release of Th1 cytokines and may downregulate Th2 inflammation at the same time. Interestingly, decreased expression of TLR9 has been demonstrated in sinonasal epithelial cells (SNEC) from patients with recalcitrant CRSwNP, which could indicate that a deficient Th1 response in these patients contributes to Th2 skewed inflammation in CRSwNP. Furthermore, IL-33, a mediator produced by SNEC that activates the production of pro-Th2 mediators from ILCs, has been shown to have 3 times higher levels of mRNA expression in treatment-resistant CRSwNP than treatment-responsive CRSwNP. These studies support the concept that innate immune defects contribute to the more recalcitrant forms of CRSwNP and that the resistance to treatment may relate to dysregulated mucosal immunity.
Genetics of Innate Immunity in Chronic Rhinosinusitis
Single-nucleotide polymorphisms (SNPs) are single-nucleotide variations in the DNA sequence. When an SNP occurs in a coding region of DNA, variability in the amino acid sequence can be created. There are many limitations to attempting to causally link SNPs with CRS pathogenesis. First, most genetic CRS studies have compared allele frequencies of SNPs between CRS patients and control subjects only in genes previously implicated in CRS pathogenesis. This comparison creates an inherent bias and simultaneously hinders identification of novel gene candidates. Pooling-based genome-wide association scans (GWAS) allow unbiased investigation of the human genome with analysis of hundreds of thousands of possible candidate SNPs at one time. Bosse and colleagues identified nearly 600 SNPs from 445 genes potentially associated with CRS. Given the multifactorial nature of CRS, the effect size of genes relevant to CRS are likely to be small, necessitating large sample sizes to achieve sufficient statistical power. Most studies have been underpowered and may be confounded by genetically diverse patient populations. Even when an SNP is associated with CRS, this does not automatically imply direct causation through gene dysregulation. Some SNPs may represent unrelated markers genetically linked to an unknown causal genetic variation, whereas others may have indirect roles by affecting transcription of undetermined genes truly involved in CRS. In genome-wide comparisons, implementing statistical corrections for multiple testing is critical in order to avoid false positive associations. When associations are replicated in multiple studies, confidence grows that a true relationship exists.
Because genetic variation in innate immunity genes appears to be a promising target for studies in CRS, multiple investigations have been attempted, but to date, definitive relationships have not been demonstrated. Only a few associations have been replicated, most notably those mentioned above, and causal associations remain unproven. Other than the studies of patients with well-characterized defects of the bitter taste receptor TAS2R38 gene, functional assessments of identified genetic variations have not yet been reported. No association has been determined between TLRs or downstream signaling molecule SNPs and CRS, other than an unreplicated study of interleukin-1 receptor-associated kinase 4 (IRAK4) polymorphisms in Chinese patients. SNPs in the nitric oxide synthase (NOS) family of genes have also been investigated in CRS, again with few reports of unreplicated associations in limited studies. Other genes that have been implicated in individual studies include the met proto-oncogene, locus (hepatocyte growth factor receptor), and serpin peptidase inhibitor (SERPINA1), but confirmatory studies are needed.
Several SNPs for multiple innate immune genes putatively involved in CRS are summarized in Table 1 . Replication studies have been performed for acyloxyacyl hydroxylase (AOAH), the bitter taste receptor T2R38, and the Cystic Fibrosis Transmembrane Regulator (CFTR locus), and these genes are discussed below.
| Gene | Variation | Phenotype | Study |
|---|---|---|---|
| AOAH | rs4504543 | CRSsNP | Zhang et al, 2012 |
| CD14 | rs2569190 | CRSwNP | Bernstein et al, 2009, Yazdani et al, 2012 |
| IRAK4 | Multiple | CRSsNP, CRSwNP | Tewfik et al, 2009 |
| LTF | −140 A > G | CRSwNP | Zielinska-Blizniewska et al, 2012 |
| MET | Multiple | CRSwNP | Castano et al, 2010 |
| NOS1 | Multiple | Mixed population | Zhang et al, 2011 |
| NOS1AP | rs4657164 | Mixed population | Zhang et al, 2011 |
| NOS2A | Promoter VNTR | CRSwNP | Pascual et al, 2008 |
| SERPINA1 | rs1243168 | Mixed population | Kilty et al, 2010 |
| TLR2 | Multiple | CRSwNP | Sachse et al, 2010, Park et al, 2011 |
| TAS2R38 | PAV/PAV, PAV/AVI, AVI/AVI | Mixed population | Adappa et al, 2016 |
Acyloxyacyl hydroxylase
Polymorphisms in the gene for AOAH were initially associated with asthma in a GWAS in 2006. Diminished degradation of lipopolysaccharides in AOAH dysfunction leads to continuous stimulation of inflammation through a TLR-4-dependent mechanism, according to studies in AOAH knockout mice. Bosse and colleagues identified an association with an AOAH SNP rs4504543 and CRS in their GWAS. Zhang and colleagues replicated this finding and showed an association with rs4504543 and Chinese patients with CRSsNP.
Bitter taste receptors
Expression of type 2 receptor bitter taste receptors (T2Rs) has been demonstrated by cell types outside of the gustatory system, including ciliated epithelial cells and solitary chemosensory cells of the upper airway. In response to bitter stimuli, and notably quorum-sensing molecules of gram-negative bacteria, nasal epithelial cell T2R38 signaling activates release of antimicrobial peptides and nitric oxide in vitro while altering ciliary beat frequency. Tas2R38 polymorphisms have also been associated with surgical outcomes in CRSsNP.
Cystic Fibrosis Transmembrane Regulator Locus
A clear genetic link between mutations in the CFTR locus and CF, which in turn is strongly associated with CRSsNP and CRSwNP, suggests that CFTR mutations could contribute to CRS in the absence of CF. A GWAS for CRS genes by Pinto and colleagues supported a relationship between CFTR and CRS. A large series published in the Journal of the American Medical Association in 2000 established that patients with CRS were significantly more likely to be CFTR carriers than were controls. Of patients who underwent full sequencing of CFTR, 38% of patients with CRSsNP were found to have mutations. Other studies have shown much lower rates of CFTR mutations in patients with CRS; however, it is important to note that those studies only assessed the 23 most common CFTR mutations. It is thought that CFTR mutations can still affect mucociliary clearance, viscosity of mucus, and sinonasal inflammation in the carrier state.
Adaptive Immunity in Chronic Rhinosinusitis
Innate immunity plays complex roles in priming and modulating the adaptive immune response through stimulating cytokine production, driving further inflammatory response and determining T-cell differentiation, among many other actions. Although the 2 arms of the immune system have been separated in this text, in reality, they are each highly dependent on the other and intricately linked. The major cell type of the adaptive immune system, lymphocytes, are activated by binding of antigen-specific receptors to release cytokines, chemokines, and/or immunoglobulins, which in turn recruit leukocytes and orchestrate inflammatory responses. Adaptive immune responses may be characterized by the dominantly expressed cytokines into Th1, Th2, and Th17 types. The inflammation in CRS is generally mixed, although Th2 skewing with local eosinophilia is a prominent feature of CRSwNP. The major Th2-associated cytokines are IL-4, IL-5, IL-9, and IL-13. CRSsNP is more heterogeneous and less defined by a specific cytokine profile, and both forms of CRS display activation of cytotoxic T-cells, macrophages, and neutrophils. Th17 inflammatory cytokines have also been described in CRS, particularly in noneosinophilic polyps, although the role of these mediators in CRS pathogenesis is uncertain.
A study by Jyonouchi and colleagues evaluated sinonasal lavage from patients with treatment-resistant CRS (mixed types), and found elevated levels of IFN-γ, IL-5, IL-8, IL-10, and IL-18. These findings support the concept that Th1 and Th2 driven inflammation could be contributing to recalcitrant forms of CRS. Regulatory T-cell (Treg) dysfunction has also been described in CRS; however, their exact role is unclear, with conflicting reports of either Treg cell impairment or increased numbers of Tregs in CRSwNP patients. B-cells have also been implicated as contributing to CRSwNP, as evident by increased B-cell numbers as well as increased levels of chemotactic factors and cytokines that promote B-cell activation and proliferation in nasal polyps compared with tissue from CRSsNP and controls. Levels of multiple immunoglobulins have been shown to be increased in nasal polyps without a concomitant increase in the peripheral blood, suggesting local rather than systemic activation of B-cell antibody production in polyps.
Genetics of Adaptive Immune Dysfunction in Chronic Rhinosinusitis
Asthma and atopy are characterized by a Th2 inflammatory cytokine profile and tissue eosinophilia similar to CRSwNP, which has led to a search for linked genetic variants in genes encoding Th2 inflammatory mediators in these diseases. In asthma and allergy, promoter polymorphisms in IL-4 (rs2243250) and IL-13 (rs20541) have been strongly and repeatedly implicated. A small number of less powerful studies of the IL4 promoter SNPs in CRSwNP have not demonstrated a relationship, although a single Chinese study identified a CRSwNP-associated SNP elsewhere in the IL-4 gene. No association has been found between IL-13 polymorphisms and CRS. The most intriguing Th2-associated cytokine may be IL-33, which is an important stimulus for type 2 ILCs. Genetic variation in IL-33 has been implicated in allergic diseases, and an association with asthma has been well replicated. Currently, the one genetic study of IL-33 and CRS by Buysschaert and colleagues looked at more than 700 well-phenotyped Belgian patients (284 patients with CRSwNP and 427 control subjects), finding that the SNP rs3939286 in the vicinity of the IL-33 start codon was significantly associated with CRSwNP, even when controlling for atopy. Whether this SNP directly impacts the expression or function of IL-33 remains to be determined. The IL-33 receptor, ST2, has also been a target of investigation, yielding inconsistent results among studies of Canadian, Belgian, and Chinese CRSwNP patients. Other genes with unreplicated studies supporting association with CRSwNP include tumor necrosis factor-β (TNF-β), A20 (modulator of nuclear factor kB activation), IL-22 receptor a1, and 2 inhibitory receptors of IL-1. Finally, genes associated with arachidonic acid metabolism, including leukotriene C4 synthase, prostaglandin D2 receptor, and cyclooxygenase-2, have been investigated in unreplicated studies with limited study power and phenotypically heterogeneous patients.
Of the several SNPs in genes for adaptive immunity, only a few have been replicated. Other SNPs implicated in CRS but not replicated are summarized in Table 2 , and those that have been replicated are discussed in greater detail later.
| Gene | Variation | Phenotype | Study |
|---|---|---|---|
| CD8A | rs3810831 | Mixed population | Alromaih et al, 2013 |
| IL10 | rs1800629 | CRSwNP | Bernstein et al, 2009 |
| IL1A | Multiple | CRSsNP, CRSwNP | Erbek et al, 2007, Mfuna Endam et al, 2010 |
| IL1B | rs16944 | Mixed population | Erbek et al, 2007, Mfuna Endam et al, 2010 |
| IL1RL1 | Multiple | CRSsNP, CRSwNP | Castano et al, 2009 |
| ILR1N | VNTR in intron 2 | Mixed population | Cheng et al, 2006 |
| IL22RA1 | Multiple | Mixed population | Endam et al, 2009 |
| IL33 | rs3939286 | CRSwNP | Buysschaert et al, 2010 |
| IL4 | rs2243250 | CRSsNP, CRSwNP | Yea et al, 2006 |
IL1A
IL-1 is an inflammatory cytokine that activates T-cells and monocytes, induces the expression of inflammatory cytokines and proteins, and upregulates the expression of adhesion molecules. It is has been shown to be more highly expressed in nasal polyps. IL-1 exists in 2 types, IL-1a and IL-1b, and the genes encoding both are located on chromosome 2. A G-to-T base exchange SNP in exon 5 at +4845 of the IL1A gene results in an amino acid substitution of alanine to serine, possibly modifying its functionality. This SNP (rs17561) has been associated with both nasal polyposis and CRS with and without polyps in separate studies. Erbek and colleagues demonstrated a significant association with both the 4845 GT and 4845 TT genotypes and nasal polyps in Turkish patients. The 4845 GG genotype was significantly more common in controls. Mfuna Endam and colleagues studied Canadian patients with CRS (both those with and without polyps were included), demonstrating an association with rs17561 polymorphism. Interestingly, a Finnish study of asthmatics demonstrated a significant association between the 4845 GG genotype (no polymorphism present) and need for surgical nasal polypectomy. It is important to note that this study identified subjects with nasal polyps by self-report only, whereas Erbek and colleagues confirmed the presence of nasal polyps by both nasal endoscopy and CT. Mfunda Endam and colleagues identified subjects with CRS (both with and without polyps) according to the 2004 American Academy of Otolaryngology-Head and Neck Surgery Guidelines. All 3 studies were performed in geographically/ethnically (and therefore likely genetically) diverse patient populations. This factor along with differences in the identification of CRS and CRSwNP patients could explain the differing results and highlights the importance of strict definitions of CRS phenotypes. Regardless, the replication of an association with rs17561 and CRSsNP and CRSwNP is exciting and suggests the need for future study.
Tumor necrosis factor-α
TNF-α acts synergistically in the process of chronic inflammation with IL-1 to regulate the extravasation of eosinophils into polyps by inducing upregulation of vascular adhesion molecules. TNFA , the gene encoding TNF-α, is located on chromosome 6 within the class 3 region of the major histocompatibility complex. Polymorphisms of the TNFA gene are associated with chronic inflammatory diseases including asthma, and the TNFA -308 G > A (rs1800629) polymorphism is seen relatively frequently in Caucasian populations.
One SNP in the promoter region of TNFA , a G-to-A substitution at position -308, increases TNFA promoter activity because of preferential transcription factor binding to adenosine over guanine. Two separate studies found the -308 GA genotype to be associated with CRSwNP in Turkish patients. Bernstein and colleagues also found that patients with the SNP rs1800629 show an increased susceptibility to CRSwNP over controls in a US study. A Hungarian study evaluated various subgroups of CRS patients including CRSsNP, CRSwNP, CRSwNP aspirin sensitive, and CRSwNP aspirin tolerant for an association with TNFA -308 G > A. Interestingly, they found that TNFA -380 G > A was positively associated with the CRSwNP aspirin-sensitive subgroup only. All other subgroups, even those with nasal polyps who were aspirin tolerant, did not show a significant association with this polymorphism. Finally, in contradiction to the above studies, Mfunda Endam and colleagues did not find an association between any of the TNFA polymorphisms that they investigated and the development of CRS. However, this study group included CRS patients both with and without polyposis and therefore is more heterogeneous than the other studies. Overall, multiple studies have suggested an association with the TNFA -308 G > A polymorphism with CRSwNP, which warrants further investigation and may suggest a potential therapeutic target.
Immunodeficiency in Chronic Rhinosinusitis
Primary immune deficiencies (PID) are inherited disorders of immunity resulting in either dysfunctional or nonfunctional portions of the immune system. More than 120 genes have been identified that contribute to primary immunodeficiency. Any arm of the immune system (humoral, phagocytic, cellular or complement) can be affected. There are more than 200 genetic disorders affecting the immune system with an estimated prevalence of 1:2000 in the general population. Some of the more common PIDs with relevance in otolaryngology include
- •
Selective IgA deficiency
- •
IgG subclass deficiency
- •
Common variable immunodeficiency (CVID)
- •
Specific antibody deficiency
- •
Agammaglobulinemia
Common clinical manifestations of PID include recurrent infections (specifically, pneumonia, bronchiectasis, otitis media, and rhinosinusitis), unusually persistent or severe infections, and/or infections with rare organisms. PID has been demonstrated as a risk factor in the development of CRS in multiple studies, and patients with refractory CRS have higher rates of PID than the general population. In one study, an immune abnormality was identified in 55% of patients with recalcitrant CRS. CVID has been identified in 1.5% to 10% of patients with refractory CRS, whereas its incidence in the general population is thought to be around 1:50,000. In addition, evidence for a humoral immunodeficiency on testing for functional antibody response has been demonstrated in 22% to 67% of patients with recalcitrant CRS. Finally, selective immunoglobulin deficiencies (IgA, IgM and IgG3 subclass) have been demonstrated at an increased rate in this patient population.
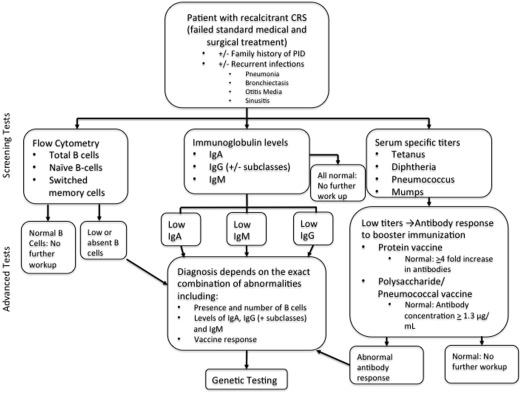
In select patients who have failed standard medical and surgical therapy, it is reasonable to consider immunologic work. Given the complexity in interpreting test results and the possible need for management of PID, immunodeficiency workups should involve an immunologist. Screening tests for humoral immunodeficiency include complete blood count (CBC) with differential, serum levels of IgA, IgM, IgG, and IgG subclasses. If the above levels are abnormal, further investigation should be undertaken with IgG titers to specific antigens for diseases that a patient has either had or been immunized to (ie, mumps, tetanus, pneumococcus, and so forth). With the exception of IgA, confirmation of humoral immunodeficiencies requires demonstration of an impaired specific antibody response. An impaired response can be demonstrated by drawing prevaccine titers followed by vaccine administration and redrawing of titers 3 to 4 weeks later; this is often done with the Pneumovax vaccine. A 4-fold increase in any vaccinated serotype previously low on titer testing should be observed. Furthermore, greater than 50% of serotypes in children and greater than 70% in adults should respond. If this testing is abnormal, then flow cytometry to count B cells or genetic testing may be indicated. Fig. 1 is a flow chart for the laboratory workup for humoral immunodeficiencies.
Workup for cellular immunodeficiency similarly begins with a CBC and absolute lymphocyte counts. Cutaneous delayed hypersensitivity testing with intradermal tests is also recommended. If either of these above tests indicates an abnormality, then further testing with T-cell flow cytometry or in vitro lymphocyte proliferation assays may be performed. Enzyme assays and a natural killer (NK) cytolysis assay complete the workup. Defects in granulocytes can be evaluated by an absolute neutrophil count and tests for neutrophil oxidative function. The workup for cellular and phagocytic immunodeficiencies is summarized in Fig. 2 .
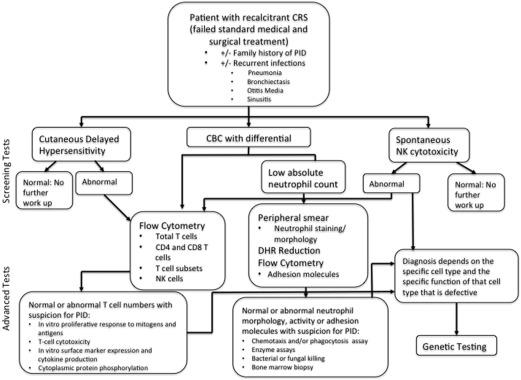
Complement function can be evaluated using 2 different assays. CH50 tests the classical complement activation pathway, whereas AH50 tests the alternative pathway. Any abnormal response can be worked up further with specific testing for components such as C3, C4, C1q, and C1 esterase inhibition. Again, at any step in the workup process, an abnormal result should prompt evaluation to an immunologist.
Recommended treatment of CRS in patients with PID includes both prophylactic antimicrobials and early culture-directed antibiotics. In addition, endoscopic sinus surgery (ESS) in patients with CRS and underlying immunodeficiency has shown similar benefits as ESS in CRS patients with normal immunity. Intravenous immunoglobulin G (IVIG) is a mechanism for inducing passive immunity in patients with immunodeficiencies. The literature regarding the impact of IVIG on sinusitis in patients with PID has been mixed. One study showed a short-term decrease in symptoms and frequency of sinopulmonary infections in patients on high-dose IVIG. Other studies have shown an increased rate of CRS in patients on IVIG. Besides an unclear benefit and possible detrimental effect on CRS symptoms in patients with PID, IVIG is associated with multiple side effects, including headache, nausea, tachycardia, shortness of breath, and anaphylaxis. Until more is understood about the impact of IVIG and CRS in patients with PID, comanagement of these patients with an immunologist and early detection and intervention of infections are appropriate.
Introduction
Chronic rhinosinusitis (CRS) is defined in adults as persistent symptoms of sinonasal inflammation for 12 or more weeks with confirmatory objective findings on nasal endoscopy or computed tomography (CT). CRS is a multifactorial disease with proposed etiopathologies including occupational/environmental exposures, infection, immune dysfunction, and genetic predisposition. As understanding of the disease evolves, clinical phenotypes have emerged that allow subclassification within the entity once identified solely as CRS. The presence or absence of polyps is used as a primary distinction because of easily recognizable clinical features and underlying inflammatory profiles. CRS with nasal polyps (CRSwNP) is characterized by T helper (Th)-2 polarization, with eosinophilia, increased levels of interleukin-4 (IL-4), IL-5, and IL-13, as well as high local production of immunoglobulin E (IgE). CRS without nasal polyps (CRSsNP) demonstrates a mixed cytokine profile that lacks Th2 polarization. High levels of interferon-γ (IFN-γ) and transforming growth factor-β have been reported in CRSsNP, but the consistency of this feature continues to be under investigation. Although Th2 polarization clearly characterizes CRSwNP in Western countries, evidence suggests that Th2 skewing may be absent in CRSwNP patients in China. This difference may be disappearing as the region undergoes modernization. A third distinct phenotype of CRS is aspirin-exacerbated respiratory disease (AERD), which is fully discussed elsewhere in this issue.
Clinically, otolaryngologists encounter refractory forms of rhinosinusitis within each phenotype. No consensus statement defining recalcitrant CRS exists; however, broadly speaking, CRS is considered refractory when it responds poorly to medical and surgical therapy. Multiple theories and possible pathophysiologic mechanisms have been proposed to explain recalcitrance in CRS. In this review, the authors focus specifically on the contribution of genetics and immunologic dysregulation.
At the most basic level, support for an underlying genetic contribution to CRS stems from observations of familial inheritance patterns. Most of these observations were made in patients with nasal polyps, and, therefore, conclusions about the familial inheritance pattern of CRSsNP cannot be drawn. In patients with nasal polyps, a heritability of 14% to 42% has been described. Cohen and colleagues subclassified CRSwNP patients into 3 groups: isolated polyps, polyps with asthma, and AERD. Interestingly, they found that AERD had the highest heritability (42%) followed by asthma and polyps (30%) and isolated polyps (15%). In addition, they demonstrated a correlation with the number of family members (frequency) with nasal polyposis and the severity of disease showing that severity is proportional to the penetrance of an underlying genetic component.
As a caveat, growing evidence suggests that multiple endotypes of CRS, yet to be fully elucidated, may be present beyond the CRSsNP/CRSwNP/AERD classification scheme. Recognition that CRS remains a complex and incompletely defined disease has important consequences for weighing the validity of genetic and immunologic studies. Without precise definitions of CRS subtypes, meaningful interpretation of research findings is difficult, particularly when detailed clinical information, including raw laboratory and radiographic findings, is not available. At present, differences among CRS subtypes are likely obscured by classifications based on an incomplete understanding, and current scoring systems used for clinical research do not capture CRS features with sufficient granularity. Because of the variety within CRS, more meticulous gathering of patient data with subclassification will almost certainly be necessary to detect true genetic and immunologic contributors. Even if precise categorization were possible, CRS fluctuates in severity and characteristics over time and can develop later in life, so flawed subtype assignments are possible, particularly in genetic studies. All of these challenges must be appreciated, and the existing CRS literature should be evaluated through this lens.
Innate Immunity and Epithelial Barrier Dysfunction in Chronic Rhinosinusitis
Among the mechanisms proposed to contribute to CRS pathogenesis and recalcitrance, many involve abnormalities of local mucosal immune defense. Sinonasal innate immune function begins with the physical epithelial barrier, mucus production, and mucociliary clearance, extending to involve secreted antimicrobial factors and phagocytic hematopoietic cells that combat infection in the airway lumen. Multiple inflammatory mediators produced by epithelial cells and other nonlymphocytic cell populations interact with the adaptive immune system to drive antigen-specific antibody production. Although lymphocytes had long been assumed to be the principal drivers of chronic inflammation, it has become increasingly apparent that tissue-bound epithelial cells, fibroblasts, macrophages, dendritic cells, mast cells, and innate lymphoid cells (ILCs) also participate in perpetuating inflammation. These cell types lack the ability to generate antigen-specific receptors through gene recombination, but instead use germline-encoded pattern recognition receptors (PRR) that bind to microbial elements and to damage-associated molecules. Genetic aberration or dysregulation of any of these innate immune pathways has the potential to contribute to CRS recalcitrance, and there is a growing body of evidence to support this concept. Abnormal mucus viscoelasticity and diminished mucociliary clearance are linked to CRS, including in genetically based diseases such as cystic fibrosis (CF) and primary ciliary dyskinesia. Alteration in expression of PRRs such as toll-like receptors (TLRs) and taste receptors may also be associated with refractory CRS.
Several antimicrobial products are present in mucus and the sinonasal mucosa and include:
- •
Acute phase proteins
- •
Neutralizing proteins
- •
Enzymes
- •
Opsonins
- •
Defensins
- •
Protease inhibitors
- •
Surfactants
Increased levels of surfactant protein A, lysozyme, C3, and C5 have been demonstrated in tissue specimens of CRSsNP compared with controls. In CRSwNP, increased levels of lysozyme, C3, cathelicidin, and acidic mammalian chitinase have been demonstrated compared with controls. Other studies have shown decreased levels of antimicrobial products in both CRSsNP and CRSwNP. Altogether, these findings indicate that abnormal functioning or regulation of the innate immune system is present in both CRSsNP and CRSwNP and may play a significant role in the persistent inflammation that is a hallmark of all CRS.
PRRs may be activated by pathogen-associated molecular patterns or damage-associated molecular patterns. These receptors are found on most cell types, including macrophages, lymphocytes, and epithelial cells. Interestingly, studies have shown that TLR2, TLR4, and TLR7 messenger RNA (mRNA) and protein levels are significantly lower in CRSsNP than controls. Similar findings of altered expression of TLR2, TLR4, TLR7, and TLR9 have been demonstrated in sinonasal tissue from patients with CRSwNP. Another class of PRRs, taste receptors, has been shown to be expressed by nasal solitary chemosensory cells and to modulate epithelial cell innate immune activity in vitro. Dysregulation of taste receptors in CRS has been suggested but not demonstrated. Although some studies have shown downregulation of PRRs and others have shown upregulation of the same PRRs, these data in aggregate imply that dysfunction or dysregulation of innate immunity is present and potentially contributing to the pathogenesis of CRS.
One example of potential innate immune dysregulation in refractory CRS is decreased expression and function of the PRR TLR9. TLR9 activation leads to the production and release of Th1 cytokines and may downregulate Th2 inflammation at the same time. Interestingly, decreased expression of TLR9 has been demonstrated in sinonasal epithelial cells (SNEC) from patients with recalcitrant CRSwNP, which could indicate that a deficient Th1 response in these patients contributes to Th2 skewed inflammation in CRSwNP. Furthermore, IL-33, a mediator produced by SNEC that activates the production of pro-Th2 mediators from ILCs, has been shown to have 3 times higher levels of mRNA expression in treatment-resistant CRSwNP than treatment-responsive CRSwNP. These studies support the concept that innate immune defects contribute to the more recalcitrant forms of CRSwNP and that the resistance to treatment may relate to dysregulated mucosal immunity.
Genetics of Innate Immunity in Chronic Rhinosinusitis
Single-nucleotide polymorphisms (SNPs) are single-nucleotide variations in the DNA sequence. When an SNP occurs in a coding region of DNA, variability in the amino acid sequence can be created. There are many limitations to attempting to causally link SNPs with CRS pathogenesis. First, most genetic CRS studies have compared allele frequencies of SNPs between CRS patients and control subjects only in genes previously implicated in CRS pathogenesis. This comparison creates an inherent bias and simultaneously hinders identification of novel gene candidates. Pooling-based genome-wide association scans (GWAS) allow unbiased investigation of the human genome with analysis of hundreds of thousands of possible candidate SNPs at one time. Bosse and colleagues identified nearly 600 SNPs from 445 genes potentially associated with CRS. Given the multifactorial nature of CRS, the effect size of genes relevant to CRS are likely to be small, necessitating large sample sizes to achieve sufficient statistical power. Most studies have been underpowered and may be confounded by genetically diverse patient populations. Even when an SNP is associated with CRS, this does not automatically imply direct causation through gene dysregulation. Some SNPs may represent unrelated markers genetically linked to an unknown causal genetic variation, whereas others may have indirect roles by affecting transcription of undetermined genes truly involved in CRS. In genome-wide comparisons, implementing statistical corrections for multiple testing is critical in order to avoid false positive associations. When associations are replicated in multiple studies, confidence grows that a true relationship exists.
Because genetic variation in innate immunity genes appears to be a promising target for studies in CRS, multiple investigations have been attempted, but to date, definitive relationships have not been demonstrated. Only a few associations have been replicated, most notably those mentioned above, and causal associations remain unproven. Other than the studies of patients with well-characterized defects of the bitter taste receptor TAS2R38 gene, functional assessments of identified genetic variations have not yet been reported. No association has been determined between TLRs or downstream signaling molecule SNPs and CRS, other than an unreplicated study of interleukin-1 receptor-associated kinase 4 (IRAK4) polymorphisms in Chinese patients. SNPs in the nitric oxide synthase (NOS) family of genes have also been investigated in CRS, again with few reports of unreplicated associations in limited studies. Other genes that have been implicated in individual studies include the met proto-oncogene, locus (hepatocyte growth factor receptor), and serpin peptidase inhibitor (SERPINA1), but confirmatory studies are needed.
Several SNPs for multiple innate immune genes putatively involved in CRS are summarized in Table 1 . Replication studies have been performed for acyloxyacyl hydroxylase (AOAH), the bitter taste receptor T2R38, and the Cystic Fibrosis Transmembrane Regulator (CFTR locus), and these genes are discussed below.



