Purpose
To describe new affected individuals of Franceschetti’s original pedigree of hereditary recurrent erosion and to classify a unique entity called Franceschetti corneal dystrophy.
Design
Observational case series.
Methods
Slit-lamp examination of 10 affected individuals was conducted. Biomicroscopic examinations were supplemented by peripheral corneal biopsy in 1 affected patient with corneal haze. Tissue was processed for light and electron microscopy and immunohistochemistry was performed. DNA analysis was carried out in 12 affected and 3 nonaffected family members.
Results
All affected individuals suffered from severe ocular pain in the first decade of life, attributable to recurrent corneal erosions. Six adult patients developed bilateral diffuse subepithelial opacifications in the central and paracentral cornea. The remaining 4 affected individuals had clear corneas in the pain-free stage of the disorder. Histologic and immunohistochemical examination of the peripheral cornea in a single patient showed a subepithelial, avascular pannus. There was negative staining with Congo red. DNA analysis excluded mutations in the transforming growth factor beta–induced (TGFBI) gene and in the tumor-associated calcium signal transducer 2 (TACSTD2) gene.
Conclusion
We have extended the pedigree of Franceschetti corneal dystrophy and elaborated its natural history on the basis of clinical examinations. A distinctive feature is the appearance of subepithelial opacities in adult life, accompanied by a decreased frequency of recurrent erosion attacks. Its clinical features appear to distinguish it from most other forms of dominantly inherited recurrent corneal erosion reported in the literature.
Recurrent corneal erosion is a condition of repeated, acute, severe eye pain attributable to defective epithelial adherence and recurrent corneal epithelial separation. The severity of the pain reflects that the cornea has one of the richest sensory innervations of the body. The condition is most commonly post-traumatic, following a glancing injury to the cornea, in which case attacks are unilateral and confined to the original site of corneal damage. In most instances, in the absence of infection, each attack recovers without significant subepithelial corneal scarring. Less commonly, the condition is bilateral and occurs either spontaneously or in response to negligible trauma. In this instance a “dystrophic” basis may be considered, although evidence of a family history is often lacking. In recent decades, in older subjects this presentation has been recognized as a manifestation of epithelial basement membrane dystrophy, wherein characteristic bilateral biomicroscopic features such as map/dot/fingerprint disorder and superficial bleb, net, and mare’s tail changes are accompanied by recurrent erosion attacks in 1 or both eyes. At least some of these are genetically determined by a mutation affecting the transforming growth factor beta–induced (TGFBI) gene. Attacks of recurrent erosion are a well-recognized feature of the phenotypically distinct, major TGFBI corneal dystrophies, as well as in Fuchs dystrophy; additionally, there have been a relatively small number of reports of inherited recurrent corneal erosion occurring as a dystrophy in its own right. All of the recurrent erosion disorders, whether traumatic or dystrophic, are presumed to be attributable to a defect in the epithelial adhesion complex, which normally ensures a tight adhesion between the basal cells of the corneal epithelium and the underlying basal lamina and Bowman layer. These attachment structures consist of the hemidesmosomes, the anchoring fibrils of type VII collagen, and the basal lamina containing collagens type IV and VII, laminin, and fibronectin.
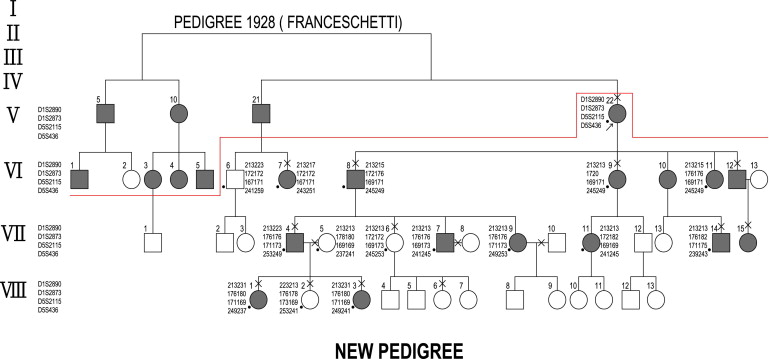
The International Committee for Classification of Corneal Dystrophies (IC3D) published a new classification in 2008 that placed each of 25 known corneal dystrophies into 1 of 4 categories based on the quality of evidence for their existence. Supportive clinical, structural, and genetic information was incorporated into a standard template for each corneal dystrophy. One template, headed “Epithelial recurrent erosion dystrophy (ERED),” included the report of Franceschetti’s “Hereditary recurrent erosion of the cornea” and the so-called “dystrophia Smolandiensis” described by Hammar and associates in a family with early-onset corneal erosion and keloid-like corneal opacifications. The study included histologic examination and DNA analysis in a large pedigree. Franceschetti, in 1928, described a separate large family with early-onset recurrent corneal erosion and autosomal dominant inheritance but, apparently, without other corneal changes. No histologic examination was performed. This 6-generation pedigree included a total of 32 affected patients, of whom only 1, the 8-year-old male proband, was clinically studied with the slit lamp. Examination revealed a corneal erosion with no other pathologic corneal findings.
The remaining 31 patients were identified as affected based on their ophthalmologic history alone. We have had the opportunity to review members of Franceschetti’s original family with recurrent corneal erosion syndrome, including newly identified members. A proportion of these were examined clinically, and in 1 case a corneal biopsy was carried out. DNA analysis was also performed.
Patients and Methods
Our proband, Patient V/22 of Franceschetti’s original pedigree, allowed us to identify new affected individuals and extend the pedigree to 8 generations ( Figure 1 ). Ten affected members of the family were interviewed and examined biomicroscopically, including careful slit-lamp examination of the cornea with and without the instillation of fluorescein. In 5 patients corneal sensitivity was measured using a Cochet-Bonnet esthesiometer. The results of these examinations are summarized in Table 1 . To assist in understanding the mechanism of the recurrent erosion attacks, a corneal biopsy was performed on Patient VI/8, a 72-year-old man, after obtaining ethical approval and with appropriate discussion and consent. A 2 × 2-mm perilimbal biopsy was performed in the left cornea in the 1 o’clock meridian, using a diamond knife to a depth of 100 μm (T.R.). The following studies were performed:
| Individual | Sex | Age(y) | Visual Acuity, OD | Visual Acuity, OS | Reduction of Corneal Sensation, OD/OS | Central Haze or Patch-like Opacification, OD/OS | Cataract, OD/OS | Cataract Extration, OD/OS | Penetrating Keratoplasty, OD/OS | AMD, OD/OS |
|---|---|---|---|---|---|---|---|---|---|---|
| V/22 | F | 101 | 20/200 | 20/200 | + | + | – | + | + | + |
| VI/7 | F | 67 | 20/32 | 20/100 | NA | + | OS+ | OD+ | − | − |
| VI/8 | M | 72 | 20/32 | 20/40 | OD-,OS+ | + | − | + | − | − |
| VI/9 | F | 70 | 20/50 | 20/40 | + | + | + | − | − | − |
| VI/12 | M | 66 | 20/25 | 20/32 | OD-,OS+ | + | + | − | − | − |
| VII/4 | M | 44 | 20/20 | 20/20 | NA | − | − | − | − | − |
| VII/14 | M | 37 | 20/40 | 20/32 | + | + | − | − | − | − |
| VII/15 | F | 31 | 20/20 | 20/20 | NA | − | − | − | − | − |
| VIII/1 | F | 12 | 20/20 | 20/20 | NA | − | − | − | − | − |
| VIII/3 | F | 7 | 20/20 | 20/20 | NA | − | − | − | − | − |
Light Microscopy and Immunohistochemistry
One-half of the specimen was fixed in 4% formaldehyde in 0.075 M phosphate buffer, dehydrated in increasing concentrations of ethanol (70%–99%), and infiltrated with paraffin (Merck, Darmstadt, Germany) at 60° C. Three-μm sections were cut, floated on de-ionized water at 45 C, and mounted as single sections on Superfrost Plus glass slides (Menzel-Glaeser, Braunschweig, Germany). Slides were stained with hematoxylin and eosin, periodic acid–Schiff (PAS), Masson trichrome, alcian blue, and Congo red, and stained immunohistochemically with polyclonal antibodies against claudin-7 (clone 5D10F3; Invitrogen, Camarillo, California, USA), E-cadherin (clone 4A2C7; Invitrogen), TGFBI, keratoepithelin-2, and keratoepithelin-15, and antibodies against decorin, which has 1 glycosaminoglycan chain of chondroitin/dermatansulfate (product number D 8428; Sigma, Roedermark, Germany). The TGFBI antibody to recombinant protein, including the full-length complementary DNA sequence (210–683) of human TGFBI, was produced in rabbits. Aminoethylcarbazole was used as a chromogen to detect the immunoreactions. The immunohistochemical staining of the specimen was compared to that of a normal postmortem cornea with a fixation time of 24 hours.
Electron Microscopy
The other half of the specimen was fixed in 4% glutaraldehyde in 0.1 M phosphate buffer (pH 7.3) for 24 hours, postfixed in 2% osmium tetroxide, processed through alcohol and propylene oxide for embedding in Araldite (Leica Microsystems, Wetzlar, Germany), and sectioned with a Reichert-Jung ultratome (Leica Microsystems). Semithin sections were stained with toluidine blue, whereas ultrathin sections were stained with uranyl acetate and lead citrate and examined with a transmission electron microscope (Zeiss, Oberkochen, Germany).
Molecular Genetic Studies
Genomic deoxyribonucleic acid (DNA) was extracted from blood samples of 15 family members and 1 spouse, and all exons of the TGFBI and tumor-associated calcium signal transducer 2 (TACSTD2) genes whose mutations are responsible, respectively, for the TGFBI-related corneal dystrophies and gelatinous drop-like corneal dystrophy were sequenced (F.L.M. and D.F.S.). Intron-exon boundaries were sequenced as described previously. In addition, the following flanking microsatellite markers were analyzed and haplotypes were constructed as described: D5S2115 and D5S436 (TGFBI); D1S2890 and D1S230 (TACSTD2).
Results
The extended pedigree is shown in Figure 1 . All affected individuals described the early onset of severe attacks of eye pain attributable to corneal erosions in 1 or the other eye, associated with a red eye, epiphora, and photophobia. The earliest onset was at 5 years and the latest at 9 years. The disease demonstrated an autosomal dominant inheritance pattern with several examples of male-to-male transmission. Ten affected individuals (V/22, VI/7, VI/8, VI/9, VI/12, VII/4, VII/14, VII/15, VIII/1, VIII/3) and 3 nonaffected family members (VII/6, VIII/2, VIII/6) were examined at the slit lamp ( Table 1 ) and examples of the lesions were photographed ( Figure 2 , Top; Supplemental Figures 1 and 2 , available at AJO.com ).
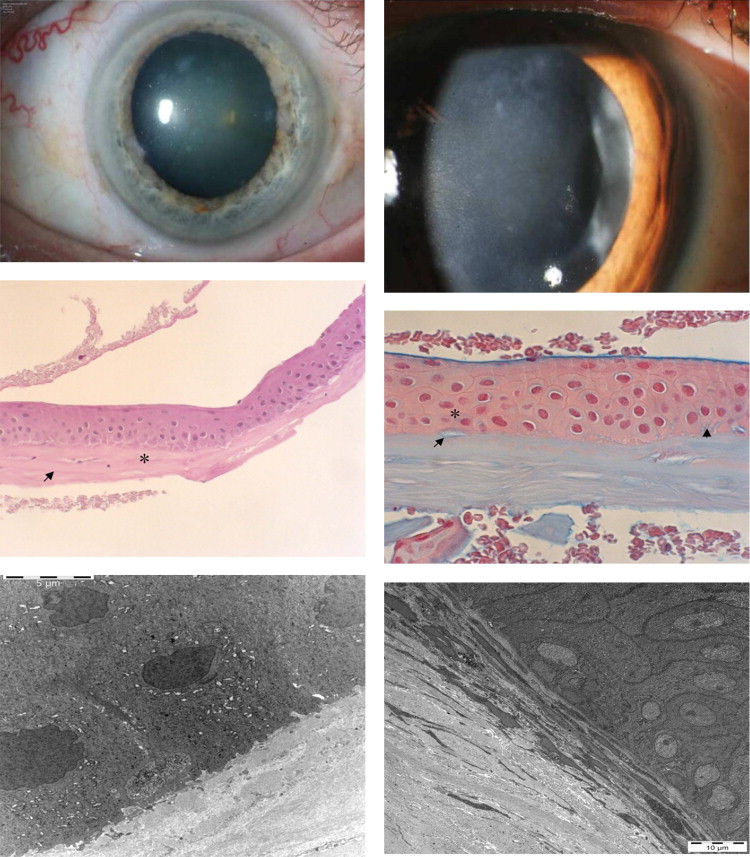
A 12-year-old female patient (VIII/1) with a history of attacks in each eye from the age of 5 suffered from ocular pain when she was examined at the slit lamp. At this time she showed bilateral punctate epithelial defects without other corneal changes. Attacks often occurred during the second half of the night and lasted for hours or even days. The clinical histories showed that young patients were frequently kept home from school because of the severe ocular discomfort. Although ophthalmic examination was critical for diagnosis, healing of the epithelium prior to examination caused some parents to incorrectly assume that their affected children were simulating pain in order to avoid school. The use of extended-wear contact lenses markedly reduced the subjective symptoms, as demonstrated in this 12-year-old girl and also in an affected adult (VII/15) who had completely clear corneas in the pain-free stage ( Supplemental Figure 3 , available at AJO.com ). A typical sequence of events was reported by a 42-year-old patient (VII/4) who also showed bilateral clear corneas in the pain-free stage of his condition, in spite of a history of recurrent, early-onset epithelial erosions. His first attack of ocular pain occurred at the age of 5 years and affected predominantly the right eye. Severe ocular pain attacks lasted during childhood and persisted until the age of 20. After age 30 the attacks occurred only 1 or 2 times per year. The pattern of symptoms in Patient VII/4 exemplifies that in all other affected adults of Franceschetti’s family. The individuals VII/4, VII/15, VIII/1, and VIII/3, who were between 7 and 44 years of age, were asymptomatic at the time of examination and had clear corneas, without fluorescein staining, despite numerous previous erosive attacks ( Table 1 ; Supplemental Figure 3 , available at AJO.com ). Slit-lamp examination of Patients VI/7, VI/8, VI/9, VI/12, and VII/14, who were between 37 and 72 years of age, showed bilateral central haze consisting of diffuse epithelial/subepithelial and patch-like opacities associated with visual impairment ( Table 1 ; Figure 2 , Top; Supplemental Figures 1 and 2 , available at AJO.com ). Confocal microscopy of Patient VI/12 disclosed irregular, diffuse opacities at the level of the corneal epithelium ( Supplemental Figure 4 , available at AJO.com ). Corneal sensation measured with the Cochet-Bonnet esthesiometer (normal value: 5.0 cm) was reduced in Patient VI/9 to right/left 0.0 cm/2.0 cm and in Patient VII/14 to right/left 1.5 cm/1.5 cm. The 101-year-old female proband (V/22), with a follow-up of 80 years, showed bilateral clear grafts after penetrating keratoplasty performed abroad 12 years previously ( Table 1 ; Supplemental Figure 5 , available at AJO.com ). The residual host cornea showed diffuse subepithelial opacities. Unfortunately, no histologic examinations of the corneal buttons were performed. It was not possible for us to get material of the specimen blocks of Patient V/22. Family members VI/10, VI/11, VII/7, VII/9, and VII/11 were considered affected based on family history.
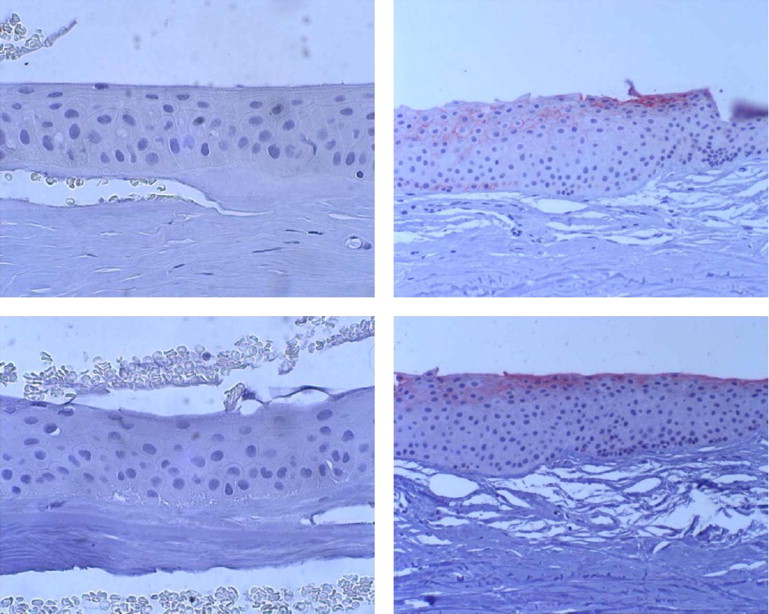
Light microscopy of biopsy from Patient VI/8 ( Figure 2 , Middle) showed an irregular basal epithelium with enlarged intercellular spaces. Alcian blue–positive deposits were present both intracellularly and intercellularly. The Bowman layer was partially destroyed and an avascular connective tissue pannus was present between the basal epithelium and Bowman layer. Congo red staining was negative.
Electron microscopy ( Figure 2 , Bottom; Supplemental Figures 6 and 7 , available at AJO.com ) showed irregularity in size and shape of the basal epithelial cells and, as already found in conventional histology, enlarged intercellular clefts containing deposits, which corresponded to the alcian blue–positive ones. There was also pannus tissue containing numerous fibrocytes.
Immunohistochemistry
In the specimen studied here, there was a lack of expression of both the tight junctional protein claudin 7 and E-cadherin, a component of the desmosome ( Figure 3 , Top and Bottom). There was positive staining for keratoepithelin-2 and keratoepithelin-15 in basal epithelium and basal membrane ( Supplemental Figures 8 and 9 , available at AJO.com ), more than in the normal postmortem cornea, where only slight staining was seen in few basal epithelial cells. Decorin expression appears to be enhanced in the basal epithelial layer compared to the normal postmortem cornea ( Supplemental Figures 10 and 11 , available at AJO.com ).