DESCRIPTION
Keratoconus is a clinical term used to describe a condition in which the cornea assumes a conical shape as a result of noninflammatory thinning. The corneal thinning in keratoconus induces irregular astigmatism, myopia, and protrusion, resulting in mild to marked impairment in the quality of vision (
1,
2). It is a progressive disorder ultimately affecting both eyes, although only one eye may be affected at the initial encounter with the patient (
3,
4).
Keratoconus classically has its onset at puberty and is progressive until the third to fourth decade of life, when it usually arrests. It may, however, commence later in life and progress or arrest at any age. It is most commonly an isolated condition, although it is frequently linked with other disorders in isolated cases (
1).
Commonly reported associations include Down’s syndrome, Leber’s congenital amaurosis, and connective tissue disorders. Several reports have shown a high incidence of mitral valve prolapse (58%) in patients with advanced keratoconus (
3,
5). Atopy, eye rubbing, and hard contact lenses are also reported to be highly associated with this disorder. From 6% to 14% of cases reported to date have a positive family history or show evidence of familial transmission (
1,
2,
3,
6).
The incidence of keratoconus is variable, with most estimates ranging between 50 and 230 per 100,000 (approximately 1 in 2,000), with a prevalence rate of 54.5 per 100,000 (
1,
7,
8,
9,
10). The variability in the reported incidence reflects the subjective criteria often used to establish the diagnosis, with subtle forms often being overlooked. Keratoconus occurs in all ethnic groups, with no male or female preponderance.
SYMPTOMS AND SIGNS
Symptoms are highly variable and are in part dependent on the stage of the disorder. Early in the disease there may be no symptoms, and the only sign may be an inability to refract the patient to a clear 20/20. In advanced disease there is significant distortion of vision accompanied by a profound decrease in vision (
1,
2). Patients with keratoconus fortunately never become totally blind as a result of their corneal disease.
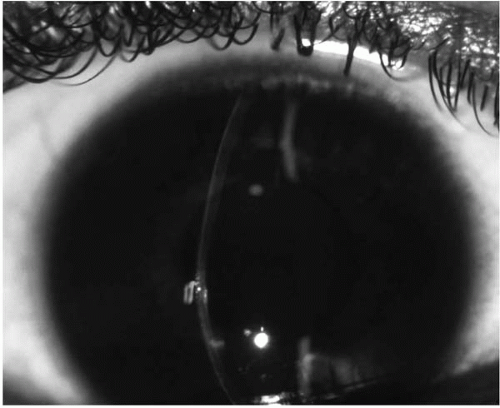
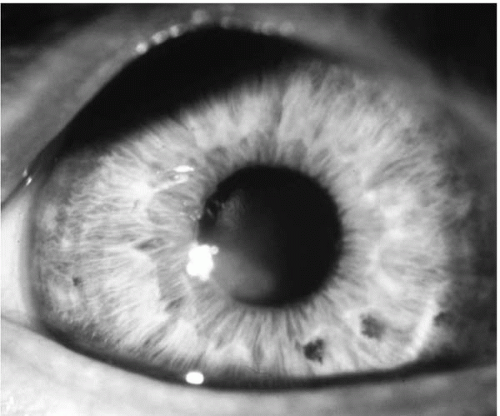
Clinical signs also differ depending on the severity of the disease at the time of presentation (
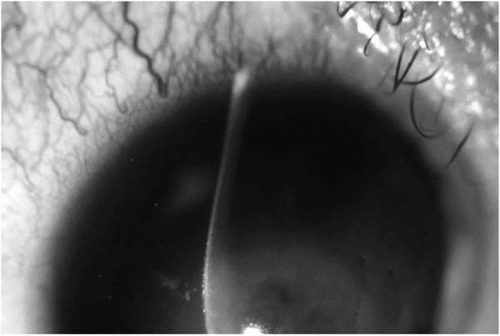
Table 49-1). In moderate to advanced disease, any one or combination of the following signs may be detectable by slit-lamp examination of the cornea: conical protrusion (
Fig. 49-1), stromal thinning (centrally or paracentrally) (
Figs. 49-2 and
49-3), an iron line partially or completely surrounding the cone (Fleischer’s ring), and Vogt’s striae, which are fine vertical lines in the deep stroma and Descemet’s membrane that parallel the axis of the cone and disappear transiently on gentle digital pressure (
Figs. 49-4 and
49-5). Other accompanying signs can include epithelial nebulae, anterior stromal scars (
Fig. 49-6), enlarged corneal nerves, increased intensity of the corneal endothelial reflex, and the presence of subepithelial fibrillary lines (
2,
3,
11).
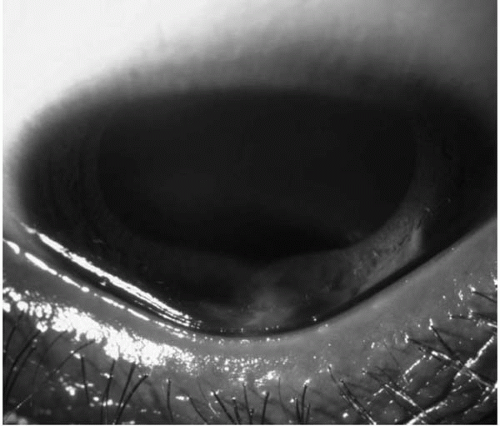
Munson’s sign and Rizutti’s sign are useful external signs associated with keratoconus (
11). Munson’s sign is a V-shaped conformation of the lower lid produced by the ectatic cornea in downgaze (
Fig. 49-7). Rizutti’s sign is a sharply focused beam of light near the nasal limbus, produced by lateral illumination of the cornea in patients with advanced keratoconus.
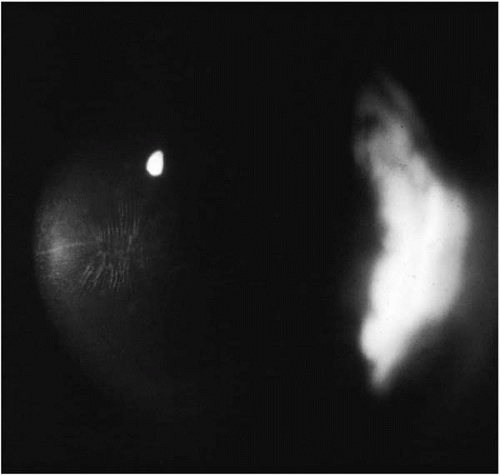
Early in the disease the cornea may appear normal on slit-lamp biomicroscopy. However, there may be slight distortion or steepening of keratometry mires centrally or inferiorly. In such instances it is useful to dilate the pupil. Retroillumination may reveal scissoring of the retinoscopic reflex or the “Charleaux” oil droplet sign, which may confirm the diagnosis in suspicious cases (
12). In these early cases, where the cornea appears normal but keratoconus is suspected, videokeratography of the cornea is the best means for confirming the diagnosis (
1).
Ultrasonic pachymetry may be used to document corneal thinning in patients with suspected keratoconus. It cannot be solely relied on to make the diagnosis because of
the large range and variation of pachymetry readings both centrally and paracentrally in the normal population (
13).
TOPOGRAPHIC DIAGNOSIS
Several devices are currently available for detecting keratoconus by measuring the anterior corneal topography. These range from simple inexpensive devices such as a hand-held keratoscopes (Placido disks) to expensive sophisticated devices such as computer-assisted videokeratoscopes. With hand-held keratoscopes, such as the Klein keratoscope, early keratoconus is characterized by a downward deviation of the horizontal axis of the Placido disk reflection (
14,
15). In the past, nine-ring photokeratoscopes, such as the Corneascope (Kera Corp., Santa Clara, CA) were commonly used by cornea specialists. With this device early keratoconus is characterized by compression of the mires inferiorly or inferotemporally (
16).
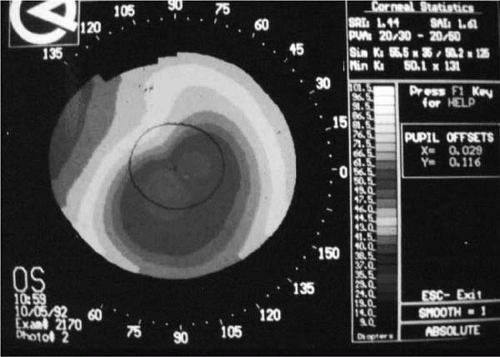
Computer-assisted videokeratoscopes that generate color-coded maps and topographic indices are currently the most sensitive technique for confirming the diagnosis of keratoconus. Videokeratography in keratoconus has three characteristic features: an increased area of corneal
power surrounded by concentric areas of decreasing power, inferior-superior power asymmetry, and skewing of the steepest radial axes above and below the horizontal meridian (
Fig. 49-8).
Several studies have characterized the topographic patterns of clinically detectable keratoconus (
17,
18). The pattern found is usually the same in the two eyes of an individual patient, although it may be more advanced in one eye relative to the other.
The majority of patients have peripheral cones, with steepening extending into the periphery. In this group, corneal steepening is usually confined to one or two quadrants. A smaller group of patients has central topographic alterations. Many central cones have a bow-tie configuration similar to that found in naturally occurring astigmatism. In the keratoconus patient, however, the bow-tie pattern is asymmetric, with the inferior loop being larger in the majority of cases. In contrast to patients who have with-the-rule astigmatism, in keratoconus the steep radial axes above and below the horizontal meridian appear skewed, giving the bow-tie a lazy-eight configuration. Another pattern found in central cones is more symmetric steepening without a bow-tie appearance. These peripheral and central cones probably correspond roughly to the oval-sagging and nipple-shaped cones described by Perry et al. (
19).
Similar patterns have been noted in clinically normal family members of patients with keratoconus and in the clinically normal fellow eye of patients with clinically unilateral keratoconus (
20,
21,
22). These patterns are less pronounced (as measured by dioptric power) than the patterns noted in clinically obvious keratoconus (
20,
21).
HISTOPATHOLOGY
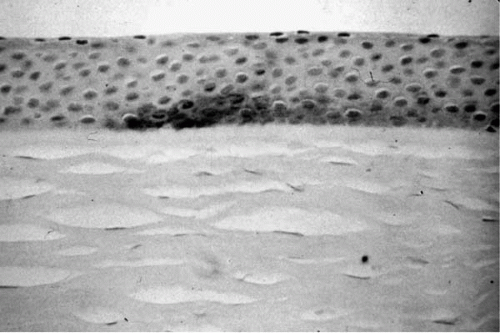
Thinning of the corneal stroma, breaks in Bowman’s membrane, and deposition of iron in the basal layers of the corneal epithelium (
Fig. 49-9) are the classical histopathologic features of keratoconus. Depending on the stage of
the disease, every layer of the cornea can become involved in the pathologic process. Fine details of these processes are most clearly appreciated by electron microscopy.
The epithelium may show degeneration of its basal cells, breaks within and downgrowth of epithelium into Bowman’s membrane, particles within a thickened subepithelial basement membrane-like layer and between basal epithelial cells, and accumulation of ferritin particles within and between epithelial cells most prominently in the basal layer of the epithelium. Changes in Bowman’s layer may include breaks filled by eruptions of underlying stromal collagen, periodic acid-Schiff (PAS)-positive nodules, Z-shaped interruptions possibly due to separation of collagen bundles, and reticular scarring. Compaction and derangement of fibrillar architecture in the anterior stroma may occur, along with a decrease in the number of collagen lamellae. Normal and degenerating fibroblasts and keratocytes may be seen. A fine granular and microfibrillar material may be associated with the keratocytes (
2).
Descemet’s membrane is rarely affected except for breaks seen in acute hydrops. The corneal endothelium is also usually normal. Reported endothelial abnormalities include intracellular “dark structures,” pleomorphism, and elongation of cells with their long axis toward the cone.
Gross histopathologic analysis of corneal buttons from patients undergoing penetrating keratoplasty for keratoconus has revealed the presence of two types of cone morphology: nipple-type cones, which are located centrally, and ovaltype (sagging) cones, which are located inferiorly or inferotemporally (
19).
Histopathologic examination of corneal buttons in patients with a history of acute hydrops reveals stromal edema (
Fig. 49-10). During acute hydrops, Descemet’s membrane separates from the posterior corneal surface and retracts into scrolls, ledges, or ridges. During the repair process, corneal endothelium extends over the anterior and posterior surfaces of the detached Descemet’s membrane and denuded stroma. Endothelial integrity is usually reestablished 3 to 4 months after the acute event (
23).
ASSOCIATED DISORDERS
Keratoconus has been reported as an isolated sporadic disorder, and in association with other rare genetic disorders, Down’s syndrome, Leber’s congenital amaurosis, and connective tissue disorders. It has also been associated with hard contact lens wear and eye rubbing. Small but significant proportions of patients have a positive family history of the disorder (
2,
24,
25,
26) (
Table 49-2). These associations, however, require further critical evaluation.
By far the most common presentation of keratoconus is as an isolated sporadic disorder with no other systemic or ocular disease detectable by clinical evaluation. To put this in perspective, of 1,200 consecutive keratoconus patients screened in a genetic research study at the Cedars-Sinai Medical Center in Los Angeles, 98% had keratoconus with no associated genetic disease.
A list of reported associations with keratoconus is presented in
Table 49-3. For the most part these associations should be regarded to have occurred by chance. For example, if the incidence of keratoconus is 1 in 2,000 and the incidence of neurofibromatosis is 1 in 4,000, then there is a 1 in 8,000,000 chance that these two disorders could occur together by chance alone (30 potential cases in the United States). Rare associations with keratoconus are important, however, particularly if they occur as a result of a chromosomal translocation and the disorder cosegregates with keratoconus. This may provide clues as to the chromosomal location of the inherited form of keratoconus. As such, it is worthwhile to perform cytogenetic studies in patients with keratoconus who have mental retardation or other rare known genetic disorders that result from chromosomal translocations.
Down’s syndrome has been reported to have a high association with keratoconus. The incidence of keratoconus in patients with Down’s syndrome ranges from 0.5% to 15% (
2,
27,
28). This is 10 to 300 times more frequent than in the general population. Similarly, a high incidence of keratoconus has been reported in patients with Leber’s congenital amaurosis, with up to 30% of patients with amaurosis above the age of 15 manifesting keratoconus (
29). Both of
these associations, however, have been attributed to a higher incidence of eye rubbing in these two disorders (due to increased blepharitis in Down’s syndrome and the oculodigital sign in Leber’s congenital amaurosis). Elder’s (
30) study of children in a school for the blind contradicts this theory and suggests the association with keratoconus might be due to genetic factors rather than eye rubbing.
Several reports suggest an association between keratoconus and connective tissue disorders (
31,
32,
33,
34,
35). Case reports have linked keratoconus with disorders of collagen metabolism such as osteogenesis imperfecta and subtypes of Ehlers-Danlos. Another study reported joint hypermobility in 22 of 44 (50%) of keratoconus patients. Two recent studies dispute the association of joint hypermobility and keratoconus (
36,
37). In the latter study, 34/218 (16%) of keratoconus versus 10/183 (5%) of normal age-matched controls had joint hypermobility (not statistically significant (
Table 49-4). Other compelling evidence in support of a connective tissue abnormality in keratoconus does exist, however. Several reports have suggested an association between advanced keratoconus and mitral valve prolapse (
38,
39). In one study, mitral valve prolapse was found in 58% of keratoconus patients requiring surgery and only 7% of normal controls (
39).
Many studies report a high association of eye rubbing in patients with keratoconus, but a cause-and-effect relationship is difficult to prove (
2). A preliminary study suggests that keratoconus patients do rub their eyes more often than normal controls (
Table 49-4) (
37).
Another form of mechanical trauma implicated in the pathogenesis of keratoconus is caused by contact lenses (
2,
40,
41). In early keratoconus, patients have mild myopic
astigmatism with clinically normal corneas. Vision is typically best corrected with rigid contact lenses. Thus, it is extremely difficult to determine which came first, the keratoconus or the contact lens wear. In none of the reports citing these associations were topographic studies done prior to contact lens fitting. There is no way to determine whether these patients had early keratoconus prior to wearing contact lenses. However, it is theoretically possible that mechanical trauma induced by eye rubbing and or hard contact lens wear might act as environmental factors enhancing the progression of the disorder in genetically predisposed individuals.
Although atopy is often cited as being highly associated with keratoconus, a review of the literature suggests conflicting data regarding this association (
2,
42,
43,
44). In a study conducted at Cedars-Sinai Medical Center in Los Angeles, 96/218 (44%) of keratoconus patients had a history or symptoms of allergic disorders, compared with 66/183 (36%) of normal age-matched controls (
Table 49-4). This difference was not statistically significant (
37).
ETIOLOGY AND PATHOGENESIS: BIOCHEMICAL STUDIES
Despite intensive biochemical investigation into the pathogenesis of keratoconus, its etiologic basis remains poorly understood. Corneal thinning appears to result from loss of structural components in the cornea, but why this occurs is not clear. Theoretically the cornea can thin for the following reasons: fewer collagen lamellae, less collagen fibrils per lamella, closer packing of collagen fibrils, or various combinations of the above. This may result from defective formation of extracellular constituents of corneal tissue, destruction of previously formed components, increased distensibility of corneal tissue with sliding collagen fibers or collagen lamellae, or a combination of these mechanisms (
45).
Biochemical and immunohistologic studies of keratoconus corneas suggest that the loss of corneal stroma after digestion by proteolytic enzymes could follow either increased levels of proteases and other catabolic enzymes (
46) or decreased levels of proteinase inhibitors (
47). Observations of the corneal proteinase inhibitors, α
1-proteinase inhibitor and α
2-macroglobulin, confer further support on the hypothesis that the degradation process may be aberrant in keratoconus (
48). Both inhibitors can be demonstrated throughout normal human corneas and corneas with diseases other than keratoconus using immunohistochemistry. However, the staining intensity in corneal epithelium from keratoconus corneas was markedly diminished. The decrease in α
2-macroglobulin was confirmed by Western dot blot assays (
48).
Tissue inhibitor of metalloproteinase-1 (TIMP-1), another proteinase inhibitor that inhibits matrix metalloproteinases, was not found to be implicated in the increased levels of gelatinolytic activity noted in prior biochemical studies of keratoconic corneas (
49,
50,
51). These proteases and inhibitors require further study to clarify their precise role in the pathogenesis of keratoconus.
Increasing attention to the communication between the corneal epithelial and stromal cells has given rise to an attractive new concept of keratoconus etiology. Wilson and co-workers (
52) have shown that the loss of anterior stromal keratocytes, which accompanies corneal epithelial abrasion or subepithelial ablation, is likely due to apoptotic cell death. Both the corneal epithelium and endothelium produce interleukin-1 (IL-1). Stromal keratocytes express the IL-1 receptor. IL-1 induces keratocyte death
in vitro, inhibits keratocyte chemotaxis, and can upregulate hepatocyte and keratinocyte growth factors (
52). It can also regulate the expression of keratocyte metalloproteinases, collagenases, and complement factors. IL-1 is postulated to be a modulator of epithelial-stromal interactions, with a role in the regulation of corneal cell proliferation, differentiation, and death. The IL-1 system has been hypothesized to play a role in the pathogenesis of keratoconus (
52). Previous work has demonstrated that keratocytes from keratoconus corneas have a fourfold greater number of IL-1 receptors than normal corneas (
53). Wilson and co-workers have suggested that the increased expression of the IL-1 receptor sensitizes the keratocytes to IL-1 released from the epithelium or endothelium, causing a loss of keratocytes through apoptosis and a decrease in stromal mass over time. This hypothesis makes sense in light of the relationship of keratoconus to eye rubbing, contact lens wear, and atopy, as the epithelial microtrauma associated within these conditions may lead to an increased release of IL-1 from the epithelium (
54).