Disorders of Neuromuscular Transmission
Normal Neuromuscular Transmission
Acetylcholine (ACh) is a natural transmitter that is synthesized chiefly in the motor nerve terminals and stored in vesicles for subsequent release. Neurotransmission begins with the release of the contents of the vesicles by exocytosis that occurs at specialized release sites located directly opposite the areas of highest concentration of ACh receptors on postsynaptic membranes (Fig. 19.1), thus minimizing the distance that the transmitter must travel to reach the receptor site. When ACh combines with its receptor, a transient increase of permeability to sodium and potassium ions occurs, resulting in membrane depolarization. When a nerve impulse arrives at the motor nerve terminal, the amplitude of the depolarization normally is sufficient to trigger an action potential that is propagated along the muscle membrane. The muscle action potential, in turn, initiates the sequence of events that leads to muscle contraction. The entire process is rapid, on the order of a millisecond. It is terminated by the removal of ACh, in part by its diffusion away from the neuromuscular junction (NMJ), but mostly by the action of acetylcholinesterase, which rapidly hydrolyzes ACh.
Myasthenia Gravis
Myasthenia gravis (MG) is a disease characterized clinically by muscle weakness and fatigability. It is caused by a reduction in the number of available ACh receptors at NMJs. Receptor depletion is mediated by one or more antibodies directed against ACh receptors or other NMJ postsynaptic membrane constituents, resulting in impaired neuromuscular transmission. Such antibodies, which either degrade or block the receptors, are found in the serum of 80% to 90% of patients with MG, although some patients with MG who have no antireceptor antibodies have an immunoglobulin (Ig) G antibody that binds to muscle specific kinase (MuSK). Finally, about 9% of MG patients are seronegative for both anti-ACh receptor antibodies and anti-MuSK antibodies. These doubly seronegative patients often have purely ocular disease.
Epidemiology
Autoimmune MG affects all races and ages, with an incidence of 4 to 5 per 100,000. There is a female predominance overall, but the sex predilection is age dependent, with women predominating among younger patients and men among those who are older at diagnosis.
Ocular Versus Generalized MG
In about 60% to 70% of patients, MG first or only affects the extraocular muscles, levator palpebrae superioris, orbicularis oculi, or a combination of these. Patients in whom only the ocular muscles are affected are said to have “ocular” MG. Patients in whom nonocular muscles initially are affected or are affected at the same time as the ocular muscles, or who develop nonocular muscle involvement after initial ocular muscle involvement are said to have “generalized” MG. In fact, the vast majority of patients (>90%) with “ocular” MG will, usually within 18 to 24 months after the onset of ocular signs, develop evidence of “generalized” disease. Thus, patients in whom “ocular” MG has been diagnosed on the basis of isolated ocular manifestations should be
warned about the tendency for generalization, and the symptoms of generalization (see below) reviewed with them. There is some suggestion that treatment of patients with ocular MG with systemic corticosteroids decreases the likelihood of subsequent development of generalized MG (see below).
warned about the tendency for generalization, and the symptoms of generalization (see below) reviewed with them. There is some suggestion that treatment of patients with ocular MG with systemic corticosteroids decreases the likelihood of subsequent development of generalized MG (see below).
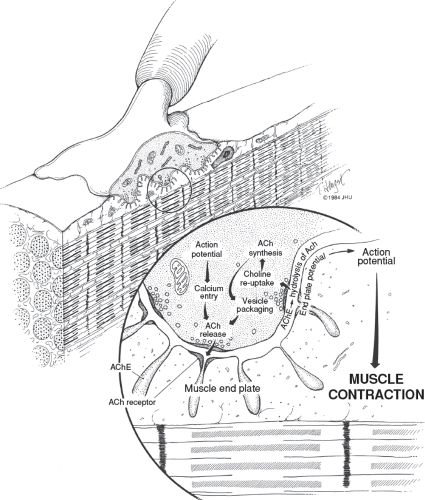 Figure 19.1 Drawing of the normal neuromuscular junction. Note the sites of acetylcholine (ACh) release, the location of acetylcholine receptors, and the location of acetylcholinesterase (AChE). |
Ocular Signs
The clinical hallmark of both ocular and generalized MG is variable weakness of the affected muscles. The weakness varies from day to day and even from hour to hour, but typically increases toward evening. Transient weakness is often associated with physical exertion.
Affected muscles fatigue if contraction is maintained or repeated. In patients with ocular MG, the most commonly affected muscles being the levator palpebrae superioris, the extraocular muscles, and the orbicularis oculi. Indeed, as noted above, the levator palpebrae superioris and extraocular muscles are affected initially in 60% to 70% of cases, and these muscles eventually are affected in over 90% of patients. When weakness of these muscles is combined with weakness of the orbicularis oculi, the combination is highly suggestive of MG.
Affected muscles fatigue if contraction is maintained or repeated. In patients with ocular MG, the most commonly affected muscles being the levator palpebrae superioris, the extraocular muscles, and the orbicularis oculi. Indeed, as noted above, the levator palpebrae superioris and extraocular muscles are affected initially in 60% to 70% of cases, and these muscles eventually are affected in over 90% of patients. When weakness of these muscles is combined with weakness of the orbicularis oculi, the combination is highly suggestive of MG.
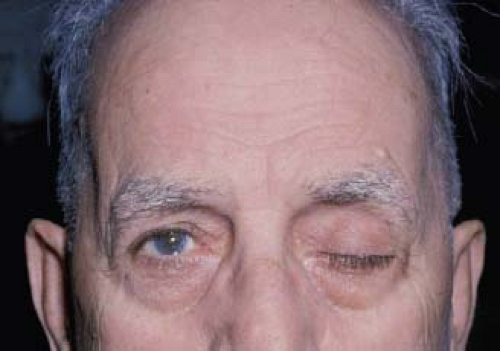 Figure 19.2 Unilateral ptosis in a 73-year-old man with myasthenia gravis. |
Ptosis and Other Signs Involving the Levator Palpebrae Superioris
Ptosis may occur as an isolated sign or in association with extraocular muscle involvement. It is characterized by its fluctuating nature and frequently shifts from one eye to the other. Ptosis often initially is unilateral but eventually almost always becomes bilateral (Fig. 19.2). When ptosis is bilateral, it may be symmetric or asymmetric, with symmetric ptosis being more common in patients who also have severe ophthalmoparesis (Fig. 19.3). The ptosis frequently is absent when the patient awakens but appears later in the day, becoming most pronounced in the evening. Repeated closure of the eyelids may make ptosis appear or may worsen it when it initially is minimal. Prolonged upward gaze also often will result in gradual lowering of the eyelids (Fig. 19.4), but fatigability of bilateral ptosis does not exclude a nonmyasthenic cause, such as dorsal midbrain compression by a mass lesion.
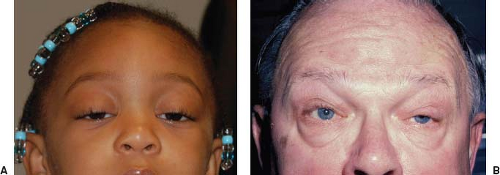 Figure 19.3 Bilateral ptosis in two patients with myasthenia gravis. A: Bilateral symmetric ptosis in a 4-year-old girl. B: Bilateral asymmetric ptosis in a 58-year-old man. |
Patients with MG-related ptosis often have worsening of ptosis on one side when the opposite eyelid is elevated and held in a fixed position (Fig. 19.5). The explanation of this “enhancement of ptosis” phenomenon is Hering law of equal innervation, which relates to the levator muscles as it does to the extraocular muscles. Manual eyelid elevation decreases the effort required for eyelid elevation ipsilaterally and thus results in relaxation of the contralateral levator and consequent worsening ptosis on that side (Video 19.1). However, enhancement of ptosis is not pathognomonic for MG. It can be seen in patients with congenital ptosis and in patients with acquired ptosis from causes other than MG. Nevertheless, in a patient with an appropriate history, the observation of enhancement of ptosis with manual elevation of the contralateral eyelid is highly suggestive of MG.
Cogan lid twitch is another important sign that suggests MG. When the patient’s eyes are directed downward for 10 to 20 seconds and the patient then is instructed to make a vertical saccade back to primary position, the upper eyelid elevates and either slowly begins to droop or else twitches several times before settling into a stable position (Video 19.2). This sign is caused by the rapid recovery and easy fatigability of myasthenic muscle.
Ophthalmoparesis and Other Abnormalities of Eye Movement
Involvement of the extraocular muscles, like ptosis, is extremely common in patients with MG, although the
reason for this is unknown. In most cases, disturbances of ocular motility and alignment are associated with ptosis; however, cases without clinical involvement of the levator muscles frequently occur. There is no set pattern to the diplopia experienced by such patients or to the nature of extraocular muscle involvement. All degrees of ocular motor dysfunction, from apparent involvement of a single isolated muscle to complete external ophthalmoplegia, occur. Thus, the abnormalities may mimic such neurologic disturbances as ocular motor nerve palsies, unilateral or bilateral internuclear ophthalmoplegia, or vertical or horizontal gaze palsies (Fig. 19.6). Unlike patients with neurologic ophthalmoparesis in whom saccadic (fast) eye movements have reduced velocities, however, patients with MG-related ophthalmoparesis have saccades with normal velocities
or velocities that are increased relative to the extent of the saccade. Even patients with MG-related complete ophthalmoplegia may show tiny “quiver movements” on attempted eccentric gaze.
reason for this is unknown. In most cases, disturbances of ocular motility and alignment are associated with ptosis; however, cases without clinical involvement of the levator muscles frequently occur. There is no set pattern to the diplopia experienced by such patients or to the nature of extraocular muscle involvement. All degrees of ocular motor dysfunction, from apparent involvement of a single isolated muscle to complete external ophthalmoplegia, occur. Thus, the abnormalities may mimic such neurologic disturbances as ocular motor nerve palsies, unilateral or bilateral internuclear ophthalmoplegia, or vertical or horizontal gaze palsies (Fig. 19.6). Unlike patients with neurologic ophthalmoparesis in whom saccadic (fast) eye movements have reduced velocities, however, patients with MG-related ophthalmoparesis have saccades with normal velocities
or velocities that are increased relative to the extent of the saccade. Even patients with MG-related complete ophthalmoplegia may show tiny “quiver movements” on attempted eccentric gaze.
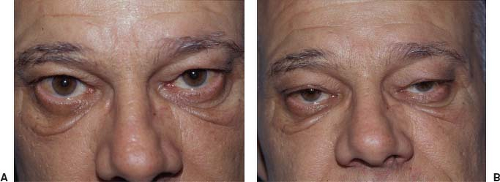 Figure 19.4 Eyelid fatigue after prolonged upgaze in a patient with myasthenia gravis. The patient was a 52-year-old man with a history of intermittent ptosis and diplopia. A: Before prolonged upgaze, the patient has minimal bilateral ptosis. B: After about 1 minute of upgaze, the patient’s ptosis is much worse. |
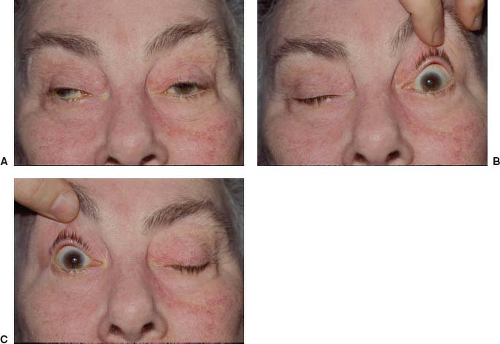 Figure 19.5 Enhancement of ptosis in a 61-year-old woman with myasthenia gravis. A: The patient has bilateral ptosis. Note also a right exotropia. B: When the left eyelid is manually elevated, the right ptosis increases. C: When the right eyelid is manually elevated, the left ptosis increases. (See also Video 19.1). |
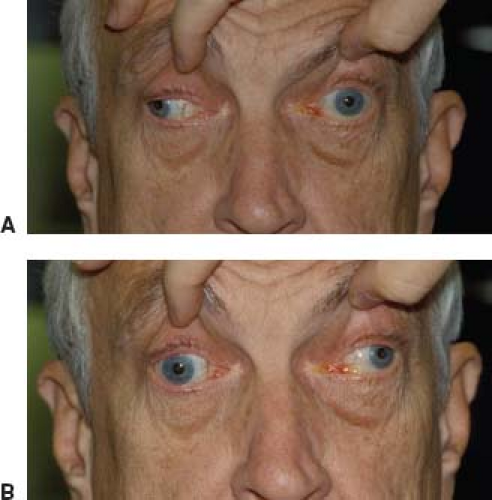 Figure 19.6 Bilateral pseudo-internuclear ophthalmoplegia in a 72-year-old man with myasthenia gravis. A: On attempted rightward gaze, the left eye adducts only to the midline. B: On attempted leftward gaze, the right eye adducts only to the midline. |
Orbicularis Oculi Involvement
Patients with MG often have involvement of the orbicularis oculi muscles that may not be apparent when ptosis or ophthalmoplegia predominates. The combination of ptosis, ocular motility disturbances, and weakness of the orbicularis oculi is found in only a few disorders, including MG, myotonic dystrophy, oculopharyngeal dystrophy, and mitochondrial myopathy. Of these, MG is by far the most common. Patients suspected of having MG therefore should have testing of orbicularis oculi strength by having the patient forcefully shut the eyes while the examiner manually attempts to open the eyelids against the forced lid closure (Fig. 19.7).
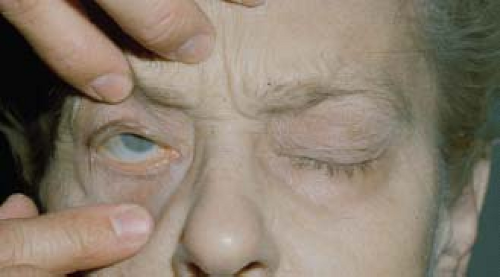 Figure 19.7 Orbicularis oculi weakness in a 78-year-old woman with myasthenia gravis. The patient is trying to close her eyes as tightly as possible; however, the closure can be overcome easily with gentle force. Note that she cannot bury the lashes on left. |
Some patients with MG exhibit a “peek” sign caused by orbicularis fatigue. In such patients, upon gentle eyelid closure, the orbicularis oculi muscle contracts, initially achieving eyelid apposition; however, the orbicularis muscle rapidly fatigues and the palpebral fissure widens, hereby exposing the sclera. The patient thus appears to “peek” at the examiner.
Pupillary Function
Patients with MG do not have clinically abnormal pupillary reactions to light or near stimulation, although subclinical involvement can sometimes be detected with pupillometry. Nevertheless, when ocular motor disturbances occur in the setting of a dilated, poorly reactive or nonreactive pupil, MG should not be considered a likely cause. Conversely, in any patient in whom an ocular motor disturbance, ptosis, or both are associated with normally reactive pupils, MG must be in the differential diagnosis.
Nonocular Signs
In patients with generalized involvement of skeletal muscles, the facies may be characteristic, showing a generalized weakness of expression. Weakness frequently affects the neck extensors and proximal limb muscles. When the muscles of expression, phonation, articulation, swallowing, and chewing are affected, there may be a characteristic facial “snarl,” dysarthric speech, nasal regurgitation of liquids, and/or the need to prop the jaw closed. When the muscles of respiration or swallowing are involved, the term “myasthenic crisis” indicates the gravity of the disease. Rarely, MG presents with respiratory insufficiency alone. On physical examination, the findings are limited entirely to the lower motor unit, without loss of reflexes or altered sensation or coordination.
Diagnostic Testing
The tests used to diagnose MG include clinical tests, assays for circulating antibodies to components of the NMJ, pharmacologic tests, repetitive nerve stimulation, and single-fiber electromyography (single-fiber EMG).
Clinical Tests
Two simple office tests are particularly helpful in supporting the clinical diagnosis of MG: the sleep test and the ice test. These tests have generally supplanted pharmacologic testing and are particularly useful in elderly or ill patients for whom pharmacologic testing may be potentially dangerous.
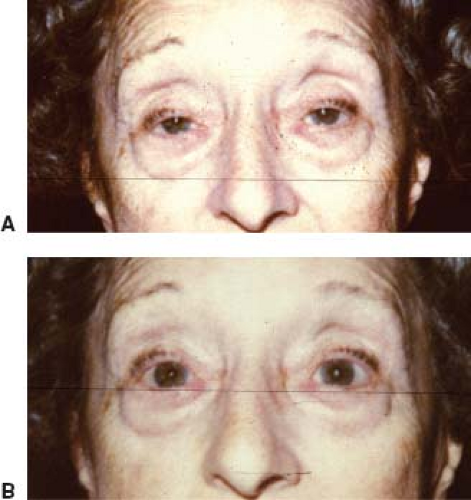
Sleep Test
The sleep test is based on the observation that when many patients with MG awaken in the morning, they have little or no ptosis or diplopia; however, these manifestations appear or worsen during the day. The sleep test is performed as follows: The patient first undergoes a complete ocular examination and external photographs. The patient then is taken to a quiet, darkened room and instructed to close the eyes and try to sleep. Thirty minutes later, the patient is awakened, immediately photographed, and measurements taken of his or her palpebral fissures, ocular alignment, and ocular motility. Most patients with MG show marked improvement in ptosis, ocular motor dysfunction, or both immediately upon awakening from sleep (Fig. 19.8). The improvement lasts 2 to 5 minutes, following which the ptosis and ophthalmoparesis recur. Patients with ptosis and ophthalmoparesis caused by disorders other than MG show no such improvement after sleep. In our opinion, the sleep test is a safe, moderately sensitive, and specific way to confirm a presumptive diagnosis of MG.