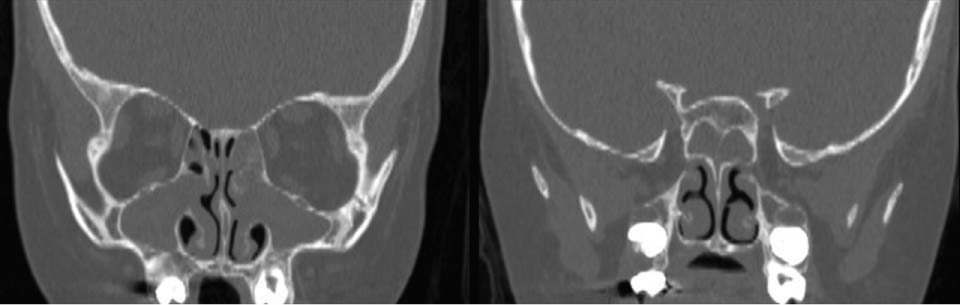
74 A 5-year-old girl with a history of cystic fibrosis (CF) was referred to a tertiary care center with several months of chronic sinusitis and increasing nasal congestion and drainage. She denied headache, fever, or facial pain. She has a history of good pulmonary function, but in the past several months, her forced expiratory volume in 1 second (FEV1) had unexplainably decreased with no improvement despite aggressive medical interventions. The patient had been previously treated with courses of Augmentin, Omnicef, and Biaxin with no improvement in her nasal congestion and FEV1. Her medical history was significant for a prior diagnosis of CF and asthma. She has not required hospitalization or intubation. Patient reported no allergies. Medications include hypertonic saline, albuterol, Flonase, Prilosec and pancrelipase. Her surgical history was negative. On physical examination a large amount of inspissated mucus was present in the middle meatus. Crusted secretions were obstructing the left anterior nose. Purulent material was seen emanating from beneath the middle turbinate. Palpation of the face was positive for mild tenderness over the maxillary sinuses. Her tympanic membranes were normal, and the examination was otherwise unremarkable. Sinus radiographs are usually abnormal children with CF. Computed tomography (CT) scans have been reported to be abnormal in 100% of those evaluated. The most likely diagnosis in this patient is chronic sinusitis with nasal polyposis. Further evaluation is needed to determine the extent of the disease and specifically to determine whether there is evidence of bony erosion from the chronic disease process. CF is an inherited autosomal recessive disorder that results in abnormal function of the exocrine glands. Although more than 800 mutations exist, the most common is a missense mutation of the CFTR gene called deltaF508. The CFTR gene codes for an essential and ubiquitous chloride channel. Exocrine glands and ducts become obstructed throughout the body as a result of the abnormal chloride channel function. This results in the most common clinical manifestations of chronic obstructive pulmonary disease and gastrointestinal malabsorption secondary to pancreatic insufficiency. Sinusitis is also common, if not universal, in patients with CF. Identified as a component of CF in the 1950s, Lurie first described the association of nasal polyposis and CF in 1957. The incidence of nasal polyposis in CF patients has been reported to be between 6 and 48%. The most common symptoms include nasal obstruction, rhinorrhea, and headache. Patients rarely complain of sinus symptoms specifically. Further the degree of disease is not well correlated with severity of symptoms. Fig. 74.1 Preoperative computed tomography in a patient with cystic fibrosis. Maxillary and sphenoid opacification. A CT scan of the paranasal sinuses was performed on this patient. As seen in Fig. 74.1 there is opacification of the maxillary and sphenoid sinuses. The osteomeatal unit is also expanded. Partial opacification of the anterior/posterior and frontal sinuses is present. Edema of the inferior turbinates is also seen. No areas of bony deficiency are identified. Chronic sinusitis with evidence in a patient with CF The effectiveness of medical management of sinusitis and nasal polyps in children with CF has been extremely disappointing. Studies evaluating the bacteriology of sinusitis in children with CF commonly identify Pseudomonas aeruginosa, Staphylococcus aureus, and Haemophilus influenzae
Cystic Fibrosis
History
Differential Diagnosis—Key Points
Test Interpretation
Diagnosis
Medical Management
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


