Corneal Endothelial Dystrophies
Patrick C. Yeh
Kathryn Colby
The posterior surface of the cornea is lined by the corneal endothelium, a monolayer of closely interdigitated hexagonal cells arranged in a mosaic (Fig. 47-1). This pattern of endothelial cells minimizes individual cell perimeter, thus reducing surface tension energy (1, 2, 3). The endothelium maintains corneal deturgescence and, thereby, corneal clarity, passively by functioning as a barrier to the influx of aqueous into the stroma and actively by pumping excess fluid out of the stroma via Na/K-adenosine triphosphatase (ATPase) pumps (4).
Human corneal endothelial cells are arrested in G1-phase and typically do not proliferate in vivo (5, 6, 7), although they can be stimulated to divide in ex vivo conditions (8). Specular microscopy, developed approximately 30 years ago (9), is currently the most commonly used technique to estimate endothelial cell density. Endothelial cell density is highest at birth (6,000 cells/mm2) (10). Cell density decreases in a nonlinear fashion during the first 2 years of life as a result of rapid growth in corneal size. Thereafter, the rate of cell loss is linear at about 0.5% to 10% per year (10,11). Average adult endothelial cell density is 2,400 cells/mm2, with a range of 1,500 to 3,500 cells/mm2 (12). Endothelial cell density is a gauge of endothelial functional reserve, although it does not always correlate with corneal thickness (13, 14, 15), suggesting that cell density is not the only determinant of endothelial cell function.
As endothelial cells die, from either normal attrition or injury, neighboring cells enlarge and migrate in an attempt to maintain the endothelial cell monolayer (16). Consequently, endothelial cells in vivo display not only a tendency toward decreasing density, but also increasing pleomorphism (variation in cell shape) and polymegethism (variation in cell size). Specular microscopy data suggest that endothelial cell density in the peripheral and paracentral cornea may be higher than central cell density (17). The peripheral endothelial cells may serve as a reservoir to repopulate the central cornea.
As endothelial cell density decreases, the risk of developing corneal edema increases (18). When the endothelial cell density decreases to a critical threshold, the cornea swells, and eventually decompensates into bullous keratopathy. Interestingly, the minimum endothelial cell density needed to maintain corneal deturgescence varies from patient to patient. Most corneas will become edematous with cell counts less than 500 cells/mm2. However, some corneas remain clear with as few as 300 cells/mm2 (15). Corneas with cell counts from 500 to 1,000 cells/mm2 are typically not edematous, but are at high risk for decompensation following intraocular surgery (19). Similarly, corneas with abnormal endothelial cell morphology are more susceptible to surgical trauma (20).
Diverse factors influence endothelial cell density and morphology. Various types of surgery including radial keratotomy (21), penetrating keratoplasty (22,23), cataract extraction (24,25), and intraocular lens implantation (26) are known to affect the endothelium. Chemical agents such as benzalkonium chloride (27) and epinephrine (28) have been shown to cause endothelial changes. Both ultraviolet (29) and infrared radiation (30), as well as the neodymium: yttrium-aluminum-garnet (Nd:YAG) laser (31), can have adverse effects on the endothelium. Endothelial changes are the hallmark of primary endothelial disorders such as cornea guttae (32) and Fuchs’ dystrophy (33). Similar changes can be secondary to diabetes (34), glaucoma (35), inflammation (36), or contact lens wear (37,38).
The corneal endothelial dystrophies encompass three major entities: (a) Fuchs’ endothelial dystrophy (FED), (b) congenital hereditary endothelial dystrophy (CHED), and (c) posterior polymorphous dystrophy (PPMD). These primary diseases of the corneal endothelium are not associated with ocular inflammation or systemic diseases. These diseases have a heritable component. These dystrophies share three characteristic features: thickening of Descemet’s membrane, disruption of normal endothelial mosaic pattern, and dysfunction of endothelial pumps (39). The diseased endothelial cells produce fibrillar collagenous tissue that is deposited between the original Descemet’s membrane and the endothelial layer. This ultrastructural change is associated with a decrease in pump function by a mechanism that is not yet understood. There may be concomitant breakdown of the passive endothelial barrier as well.
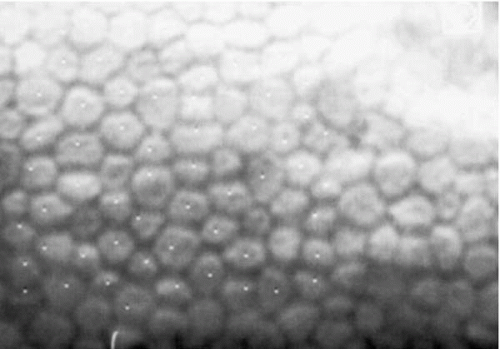 FIGURE 47-1. Specular microscopy of normal corneal endothelium. Cells are relatively uniform in size and shape. |
The corneal endothelium is derived from the neural crest, and this fact has been exploited in another classification system of the corneal endothelial dystrophies (40,41). In this scheme, the endothelial dystrophies listed above result from abnormalities in neural crest cell final differentiation. Iridocorneal endothelial (ICE) syndrome, another primary disorder of the corneal endothelium, is proposed to result from abnormal crest cell proliferation. ICE syndrome is not, strictly speaking, a dystrophy, as it is acquired and unilateral. However, ICE syndrome shares many overlapping clinical features with PPMD. Therefore, ICE syndrome is included in this chapter and considered with the classic corneal endothelial dystrophies as part of a more inclusive disease category: corneal endotheliopathies.
FUCHS’ ENDOTHELIAL DYSTROPHY
Fuchs’ endothelial dystrophy (FED) is a bilateral, slowly progressive, primary disorder of the corneal endothelium that causes corneal edema and eventually painful blindness (42). End-stage FED currently accounts for up to 29% of penetrating keratoplasties in the United States (43,44).
The disorder was first described in 1910 by the Viennese ophthalmologist, Ernst Fuchs (45), who reported 13 cases of bilateral central clouding in elderly patients, nine of whom were women. Although he initially referred to it as “dystrophia epithelialis corneae,” Fuchs speculated that “an alteration of the posterior endothelial covering” may underlie the “increased passage of the aqueous humor into the cornea.” With the newly introduced biomicroscope, Koeppe (46) noted focal “dimples” in the endothelial layer in patients with corneal edema. The term gutta (Latin: noun; pl., guttae; meaning “droplet”; adjective guttata) was first coined by Vogt (47) to describe the endothelial changes as observed under the slit lamp. In 1920, Kraupa (48) first correctly deduced the progression from the endothelial changes to epithelial edema, although others soon followed suit (49, 50, 51). Thereafter, FED was referred to as “endothelial dystrophy of the cornea.” Ultrastructural studies in the 1970s provided evidence that the pathologic changes of the epithelium and stroma in FED were the direct result of primary dysfunction of the corneal endothelium (52, 53, 54).
There is a hereditary component to FED. In our population, approximately 30% of patients have a known family history of the disease (55). An autosomal-dominant pattern of inheritance has been suggested for FED (56, 57, 58, 59), although several characteristics of the disorder make determination of the exact inheritance pattern challenging. Specifically, FED is asymptomatic in its early stages and therefore may go undiagnosed. Clinically significant manifestations of the disease occur late in life when it may be difficult to obtain complete pedigrees due to death of older family members.
Most studies suggest that FED is more common in women, with as high as a 4:1 female-to-male ratio in some series (56, 57, 58, 59). The penetrance and disease expression may vary from family to family. Women appear to be more severely affected, for unclear reasons. Epigenetic factors are likely to play a role in the expression of the disease, but these have not been well defined as of yet.
The female-to-male ratio of Fuchs’ endothelial dystrophy patients undergoing penetrating keratoplasty is about 3 to 4:1 (60,61).
The female-to-male ratio of Fuchs’ endothelial dystrophy patients undergoing penetrating keratoplasty is about 3 to 4:1 (60,61).
FED has been associated with other conditions. It may occur more frequently in patients with cardiovascular disease (62). It also has been associated with keratoconus (63,64), age-related macular degeneration (65), and short axial length with a narrow anterior chamber angle (66). The relationship between FED and open-angle glaucoma is controversial, as some studies report decreased aqueous outflow in patients with FED (67), whereas others have not (68).
Clinical Features
Fuchs’ dystrophy is a slowly progressive disorder, typically progressing over several decades. Although clinically evident in the fourth to fifth decade of life, it usually does not cause visual disturbance until patients are in their 50s or 60s. The corneal changes are usually bilateral, but often asymmetric. The disease has four stages (42,55,69).
The first stage is asymptomatic. Slit-lamp examination discloses central corneal guttae, but vision and corneal thickness are normal. Guttae, irregularly scattered excrescences in the posterior cornea, are often associated with fine pigment dusting (Fig. 47-2). Descemet’s membrane may appear opaque and thickened. At this stage, one cannot diagnose Fuchs’ dystrophy, as the vast majority of individuals with corneal guttae will not progress to corneal edema (70).
The diagnosis of FED can be made once stroma edema occurs in the setting of corneal guttae, most typically in the fifth decade of life (Fig. 47-3). In this second stage, patients have painless decreased vision, especially upon awakening. Vision may improve as the day progresses as evaporation promotes corneal deturgescence. Glare and haloes may be noted. As the disease progresses, the entire stroma becomes edematous, giving the cornea a ground-glass appearance. Specular microscopy demonstrates reduced endothelial cell density with abnormal morphology (pleomorphism and polymegathism), although advancing edema may make it difficult to obtain adequate specular images (Fig. 47-4). Ultrasonic pachymetry, which measures corneal thickness, may also be used to determine if the endothelial dysfunction is advancing (increasing pachymetry readings) or remaining stable. Normal central corneal thickness is approximately 500 to 550 μm. When pachymetry readings exceed 700 μm, the likelihood of eventual corneal decompensation requiring penetrating keratoplasty is high.
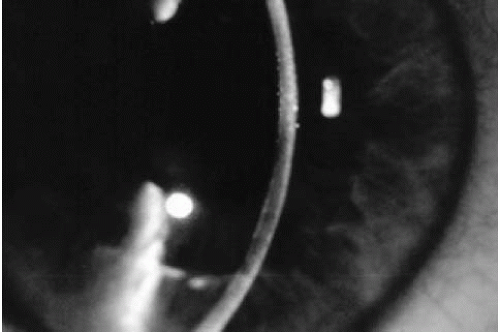 FIGURE 47-2. Central corneal guttae without stromal thickening.(see color image) |
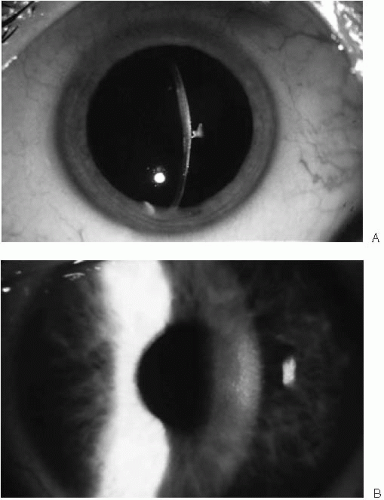 FIGURE 47-3. A: Stage 2 Fuchs’ endothelial dystrophy. Corneal guttae with pigmentary dusting create a beaten metal appearance. Note the stromal edema in central cornea; peripheral cornea is of normal thickness. B: Late stage 2 Fuchs’ endothelial dystrophy. The cornea has diffuse stromal edema with haze. No epithelial edema is noted.(see color image) |
Epithelial edema characterizes the third stage (Fig. 47-5). Initially, fine epithelial microcysts are noted. The epithelial surface is roughened, with an irregular surface texture. Vision invariably deteriorates during this stage and marked fluctuations in vision are common. Occasionally, erosive symptoms are the presenting complaint. Anterior basement membrane changes may be noted on exam. As the endothelial dysfunction worsens, epithelial cysts coalesce to form large intraepithelial and subepithelial bullae (Fig. 47-6). These bullae may rupture, resulting in
severe eye pain and rendering the patient susceptible to infection.
severe eye pain and rendering the patient susceptible to infection.
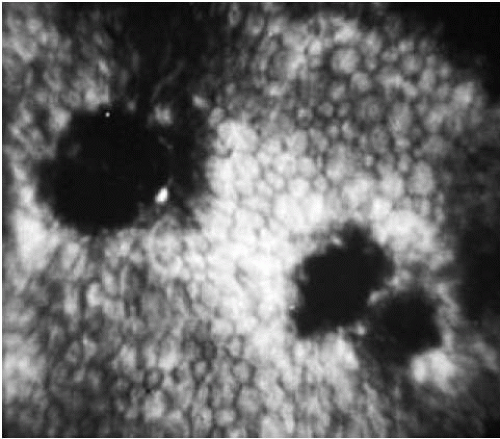 FIGURE 47-4. Specular microscopy of Fuchs’ endothelial dystrophy. The endothelial mosaic is disrupted by irregular dark areas devoid of endothelial cells (guttae). The remaining endothelial cells show increases in size (polymegathism) and variations in shape (pleomorphism). |
In the fourth stage, growth of avascular subepithelial connective tissue occurs, causing reduced vision from scarring. The cornea is opaque and compact (Fig. 47-7). Pain is decreased, but vision is severely reduced to the hand motions level. Corneal sensation is decreased or absent. With time, peripheral corneal vascularization may occur (42).
Histopathology
Normal Descemet’s membrane has two discernible layers identified by electron microscopy: an anterior banded layer and a posterior nonbanded layer. The anterior layer is only produced during fetal development. It is approximately 3 μm thick and consists of 110 nm banded collagen. The posterior nonbanded layer is deposited continuously throughout life at an average rate of about 1 to 2 μm per decade. As a result, the thickness of the posterior layer increases on average from 3 μm at age 20 to 10 μm at age 80 (71).
In FED, the anterior banded layer is normal; however, the posterior nonbanded layer is attenuated or absent (52,72). In its place is an abnormal banded layer, which thickens Descemet’s membrane to 20 μm or more (39,69). This abnormal layer consists of 110 nm collagen banded fibrils similar to those in the anterior banded layer, but which are arranged in a patchy and disorderly fashion. The abnormal posterior layer also contains spindle-shaped bundles of collagen, microfibrils, and amorphous ground material (72,73). Oxytalan fibers, not a normal constituent of the posterior cornea, are also present (74). Endothelial cells that have undergone fibroblastic transformation may be responsible for the deposition of abnormal material into the posterior layer of Descemet’s membrane (75).
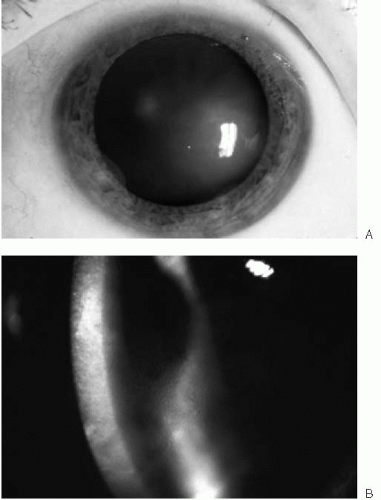 FIGURE 47-5. A: Stage 3 Fuchs’ endothelial dystrophy localized to a small area of the cornea. When endothelial cell density reaches a critical level, epithelial edema occurs, manifested by the irregular corneal light reflex. B: Stage 3 Fuchs’ endothelial dystrophy showing diffuse stromal and epithelial edema with microcystic changes.(see color image) |
Microscopically, Descemet’s membrane thickening can manifest four patterns: (a) discrete excrescences, or guttae,
protruding into the anterior chamber; (b) multilamellar excrescences; (c) excrescences buried within the multilamellar collagen tissue; or (d) multilamellar collagen tissue without excrescences (76). The excrescences cause thinning and morphologic changes in the overlying endothelial cells. As a result, the endothelial cells appear attenuated, with widely spaced nuclei, especially over the apices of the excrescences. Areas of marked thickening of Descemet’s membrane and endothelial cell attenuation have been shown histologically to correspond to areas of severe corneal edema seen clinically (73).
protruding into the anterior chamber; (b) multilamellar excrescences; (c) excrescences buried within the multilamellar collagen tissue; or (d) multilamellar collagen tissue without excrescences (76). The excrescences cause thinning and morphologic changes in the overlying endothelial cells. As a result, the endothelial cells appear attenuated, with widely spaced nuclei, especially over the apices of the excrescences. Areas of marked thickening of Descemet’s membrane and endothelial cell attenuation have been shown histologically to correspond to areas of severe corneal edema seen clinically (73).
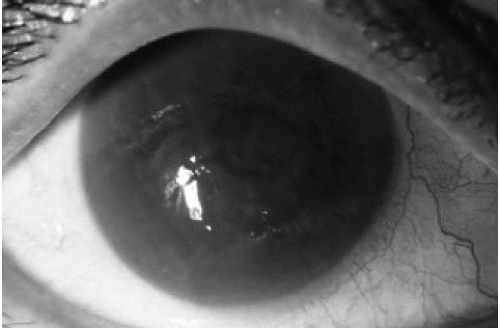 FIGURE 47-6. Bullous keratopathy. Diffuse stromal and epithelial edema are seen. The eye becomes painful with increased risk of infection when bullae rupture. |
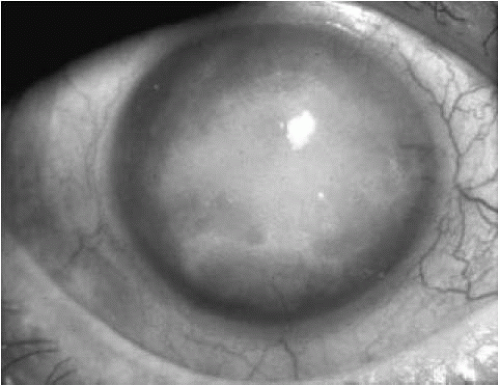 FIGURE 47-7. Stage 4 Fuchs’ endothelial dystrophy. The vision is severely diminished by the dense corneal opacification with avascular subepithelial fibrosis. However, the eye is generally comfortable. |
Ultrastructural studies of Fuchs’ dystrophy corneas have demonstrated numerous changes. Although endothelial cell morphology is largely maintained, loss of mitochondrial cristae has been noted (55). Nonspecific degenerative changes such as vacuoles, swollen organelles, myelin figures, dilated endoplasmic reticulum, clumps of pigment, and increased cytoplasmic filaments also occur (52,73,76,77). Cell size is increased to approximately 1,000 μm2, from a normal of 400 μm2. As the disease progresses, the endothelial cells lose their characteristic hexagonal shape (32). Junctional complexes between the cells are loosened, with resulting discontinuity within the endothelial cell monolayer. Scanning electron microscopy has confirmed the irregular posterior corneal surface in FED, showing severely degenerated endothelial cells with patches of cell loss exposing a fibrous network of collagen tissue underneath (75). These changes are localized to areas of corneal edema. Endothelial cells in nonedematous areas and at the corneal periphery display a relatively normal ultrastructure (73).
Specular microscopy provides in vivo assessment of endothelial cell morphology and can be used to follow the progression of FED (Fig. 47-4). Early in the disorder, specular microscopy reveals reduced endothelial count with disruption of the endothelial mosaic by hyporeflective spots. These spots, corresponding to guttae, appear dark because the endothelial cells over the excrescences are displaced posteriorly out of the plane of focus (69). When the guttae become numerous, it is usually not possible to observe the endothelial tessellation. The increasing polymegathism (increase in cell size) and pleomorphism (variability in cell shape) that characterizes progression of the disease is also easily demonstrated by specular microscopy (78).
The stroma in FED is affected secondarily as a result of the endothelial changes, but the changes in the stroma are less distinctive. In the early stages, the stroma can appear normal under the light microscope. When stromal edema ensues, there is disruption of the normal architecture of the collagen lamellae, with widened interfibrillar spaces and areas of granular filamentous material laid down by the activated keratocytes. Bowman’s layer remains intact for the most part except for occasional focal defects filled with connective tissue (53). In the advanced stage of the disease, thick, subepithelial avascular connective tissue may be observed between Bowman’s layer and the epithelial basement membrane. This fibrocellular layer contains active fibroblasts, collagen fibrils, and basement membrane-like material, and may accumulate a thickness up to 350 μm, about seven times the normal epithelial thickness (53). Vascularization of this layer may occur with time (42).
The epithelium is also passively affected as the edema spreads anteriorly. Initially, intraepithelial cysts are noted in the basal layer of the epithelium (intracellular edema). As more fluid accumulates, the cells may rupture, leading to intercellular edema. With time, the basal epithelial cells may become focally detached from the basement membrane, resulting in subepithelial bullae (53,79,80).
Pathophysiology
The underlying pathology of Fuchs’ dystrophy resides in the corneal endothelium. Abnormal production of collagenous material by the affected endothelial cells causes marked thickening of Descemet’s membrane, which becomes studded with excrescences (guttae). Changes in endothelial cell morphology and function eventually result in corneal edema.
Corneal deturgescence is primarily maintained by active transport of electrolytes (with secondary transport of water down the osmotic gradient) from the stroma to the anterior chamber via adenosine triphosphate (ATP)-requiring Na-K pumps. Both pump density (81,82) and rate (32) can be increased in early FED, potentially representing a compensatory mechanism for the decreasing number of functional endothelial cells. As the disease progresses, there is a gradual decline in the number of functional Na-K pumps (81,82). Eventually the remaining endothelial cells
can no longer maintain corneal deturgescence and corneal edema ensues.
can no longer maintain corneal deturgescence and corneal edema ensues.
The intact endothelial monolayer also serves as a passive barrier to diffusion of water and solutes from the aqueous humor. It is not clear whether this barrier function is compromised in FED. Several studies have demonstrated increased endothelial permeability in patients with central cornea guttae, suggesting that a breakdown in the endothelial barrier function may be an early finding in FED (32,83,84). However, these results were not supported by measurements made using two-dimensional scanning fluorophotometry (85).
Although there is a reasonably good understanding of the site of the primary pathology and of the natural history of FED, the etiology of its characteristic endothelial dysfunction remains an enigma. Numerous hypotheses regarding the pathogenesis of FED have been proposed, but none has as yet been proven.
A role for the fibrinolytic system in the pathogenesis of FED has been suggested, based on a report of increased fibrinogen degradation products in serum and aqueous humor of patients with FED (86). Immunofluorescence staining localized fibrinogen/fibrin antigens to abnormal Descemet’s membrane in FED, but not in normal eyes (87). Finally, a single study noted improvement in central corneal thickness and visual acuity in patients with bullous keratopathy secondary to FED treated with a systemic antifibrinolytic drug, tranexamic acid (88). Although intriguing, the significance of this finding is unclear.
Some studies have investigated the possibility that primary abnormalities in the composition of the aqueous humor may play a role in the pathogenesis of FED. Alterations in anterior chamber concentrations of multiple amino acids have been noted in FED patients (89). However, these differences may simply be a by-product of the abnormal endothelial transport mechanism.
The increased incidence and more severe phenotypic expression of FED in women have led to the speculation of a possible hormonal role in its pathogenesis (42), although there is a paucity of data supporting this hypothesis. Interestingly, though, it has been shown that the posterior nonbanded layer of Descemet’s membrane is twice as thick in normal women above age 70 as it is in age-matched men (71).
The corneal endothelium is of neuroectodermal origin. FED, along with other hereditary corneal endothelial dystrophies, may result from abnormal terminal induction or final differentiation of neural crest cells (40). Recently, missense mutations in the COL8A2 gene, which encodes the α2 chain of type VIII collagen, a component of endothelial basement membranes, were identified in familial and sporadic cases of FED (90). Similar findings in a family with PPMD led the authors to hypothesize that abnormalities in type VIII collagen may perturb the terminal differentiation of corneal endothelial cells in these diseases.
Several lines of evidence suggest that apoptosis or its regulation may play a role in FED (91,92). The average percentage of apoptotic endothelial cells is significantly higher in FED patients than in controls, although transmission electron microscopy did not show conclusive evidence of increased endothelial apoptosis in FED (91). Marked differences in keratocyte apoptosis have been found in normal and FED corneas (92). Keratocytes participate in the turnover of the extracellular matrix, and more importantly, in the maintenance of normal endothelial function by providing physical and growth factor support (93). Growth of corneal endothelial cells in culture shows a twofold increase when keratocytes are included, suggesting that keratocytes secrete factors that are involved in endothelial cell maintenance and proliferation (94). Excessive apoptosis of keratocytes, with subsequent loss of their supportive factors, could potentially represent the primary event that leads to endothelial dysfunction. The temporal relationship between keratocyte apoptosis and endothelial dysfunction has not yet been established, however.
Several lines of evidence suggest that mitochondrial abnormalities may play a role in Fuchs’ dystrophy (55). Endothelial Na-K pumps use a tremendous amount of ATP to maintain corneal deturgescence. Thus, any process that disturbs energy production or utilization will adversely affect endothelial cells function. The activity of cytochrome oxidase, a key enzyme in ATP production, is reduced in edematous areas of FED corneas, believed to reflect a decrease in either the number or the metabolic activity of endothelial mitochondria in the dysfunctional endothelial cells (95). Marked abnormalities in mitochondrial ultrastructure have been seen in corneal endothelial cells from patients with Fuchs’ dystrophy (55).
Mitochondria possess a number of unique features (96). They contain their own extrachromosomal DNA, which is inherited maternally and is distinct from nuclear DNA. The mitochondrial genome, 16.6 kilobases of circular, double-stranded DNA, codes for structural RNAs and proteins involved in oxidative phosphorylation. Mitochondria replicate, transcribe, and translate their DNA independently. Effective energy production, however, requires coordinated interactions among the mitochondrially encoded gene products and those imported from the nucleus.
The mutation rate of mitochondria DNA is 10-fold higher than that of nuclear DNA. Not only is the mitochondrial DNA bathed in the free-radical byproducts of oxidative phosphorylation, but it also lacks both protective histones and a sophisticated DNA repair system. Because mitochondrial DNA has no introns, mutations are more likely to affect coding regions. Inherited germ mutations in the mitochondrial DNA accumulate sequentially through maternal lineages. In addition to heritable mutations, nonheritable mitochondrial DNA mutations or deletions occur within somatic cells. Accumulation of somatic cell
mutations may contribute to the pathogenesis of age-related diseases such as Parkinson’s disease (55). Normal and mutant mitochondrial DNA often coexist within the same cell, a condition known as heteroplasmy. There is a threshold effect, where the proportion of mutant mitochondrial DNA required to cause clinical expression of the deficit in ATP production varies among individuals, organ systems, and within a given tissue.
mutations may contribute to the pathogenesis of age-related diseases such as Parkinson’s disease (55). Normal and mutant mitochondrial DNA often coexist within the same cell, a condition known as heteroplasmy. There is a threshold effect, where the proportion of mutant mitochondrial DNA required to cause clinical expression of the deficit in ATP production varies among individuals, organ systems, and within a given tissue.
Mitochondrial DNA abnormalities cause a diverse group of disorders, collectively referred to as the mitochondrial encephalomyopathies. These disorders, which include diseases such as Leber’s hereditary optic neuropathy and chronic progressive external ophthalmoplegia, have protean manifestations, primarily affecting cells and tissues with high energy requirements (97). The postmitotic nature of corneal endothelial cells and their tremendous metabolic demands make them a likely site to manifest mitochondrial DNA damage, either inherited or acquired. Several cases of primary corneal endothelial abnormalities in patients with mitochondrial disorders have been reported (98, 99, 100, 101). These clinical associations, combined with the unique features of mitochondrial DNA and the biologic nature of corneal endothelial cells, suggest that defects in mitochondrial DNA, either inherited or acquired, might underlie the endothelial dysfunction of Fuchs’ dystrophy (55).
Management
Early Fuchs’ dystrophy is asymptomatic and requires no treatment. Patients may wish to avoid contact lens use, as the relative hypoxia may unduly stress the compromised endothelium (102,103). Similarly, topical carbonic anhydrase inhibitors are best avoided in this population, as irreversible corneal edema has been linked to these agents (104).
Topical hyperosmotic agents, which act by temporarily increasing the osmolality of the precorneal tear film, may be used with variable success once corneal edema occurs. Some patients may benefit from using topical 5% sodium chloride solution several times upon awakening or throughout the day; ointment form may also be used prior to sleep to help reduce morning visual blurring. A hair dryer held at arms length from the cornea may also be used to facilitate corneal dehydration. Alternatively, lowering the intraocular pressure may reduce the hydrostatic pressure, which acts to push fluid into the cornea and thereby decrease corneal edema. Unfortunately, none of the currently available treatments are particularly efficacious, especially for stromal edema. In patients with bullous keratopathy and/or recurrent erosion symptoms, a loosely fit, high-water-content soft contact lens, e.g., Kontur lens, may be used to reduce the irritation and pain. Cycloplegic agents can be added to relieve the discomfort associated with ciliary spasm. There is no clear role for the use of topical corticosteroids to treat stromal edema in patients with FED (105). A conjunctival flap may reduce pain in patients with advanced bullous keratopathy who are not candidates for corneal transplantation.
Penetrating keratoplasty is indicated for patients whose reduced vision or eye pain interferes with their daily activities. Although variability exists in the literature about the long-term results after penetrating keratoplasty for FED, the graft survival and visual outcome are in general favorable. The long-term (with at least 1 year of follow-up) rate of graft clarity ranges from 61% to 98% (106, 107, 108, 109, 110, 111). Visual acuity stabilization following keratoplasty in FED is comparable to that in keratoconus and faster than that in pseudophakic or aphakic corneal edema, with 50% of patients achieved a visual acuity of better than 20/40 at 3 months (112). Nonetheless, the lifetime prognosis for corneal transplants in FED is still guarded, as progressive loss of endothelial cells always occurs and there is no source of healthy host endothelium to repopulate the graft, the way there is in keratoconus (113,114). Graft rejection, a major cause of graft failure, occurs in 5% to 29% of transplants performed for FED (108,115). Early recognition and aggressive treatment of rejection episodes can reduce the chance of ensuing graft failure (110). Patient education regarding the symptoms of graft rejection (blurred vision, eye redness, prolonged foreign-body sensation) is a key component in the management of any postkeratoplasty patient.
Many of the patients who require penetrating keratoplasty for FED are elderly and have preexisting lens opacities (107,116). Management of the cataract in FED patients undergoing keratoplasty may be approached in several ways. There appears to be no statistically significant difference in the final visual outcome between combined and nonsimultaneous procedures for FED and coexistent cataract (117). Patient age, medical health, and status of the other eye should all be considered when deciding which procedure to perform.
In patients with stage 2 Fuchs’ dystrophy with stromal edema and a visually significant cataract, phacoemulsification alone can be attempted, if the corneal view allows it. The surgeon should use liberal amounts of viscoelastic and minimal amounts of ultrasound power during cataract extraction in this setting. It is important to counsel patients preoperatively that corneal decompensation may occur following cataract surgery, but if it does not, then corneal transplantation can be delayed.
In the setting of bullous keratopathy with cataract, the decision is whether to perform a triple procedure (penetrating keratoplasty, cataract extraction, and intraocular lens implantation) or a corneal transplant alone, followed by cataract extraction after the corneal transplant has stabilized. Each approach has advantages. Simultaneous surgery allows quicker visual rehabilitation and requires only one surgery. This may be desirable for older patients and those with significant medical issues. Sequential surgeries allows for more accurate intraocular
lens power calculations (118) and allows for small incision cataract surgery, which is safer than the open-sky approach required in the setting of a triple procedure for advanced bullous keratopathy.
lens power calculations (118) and allows for small incision cataract surgery, which is safer than the open-sky approach required in the setting of a triple procedure for advanced bullous keratopathy.
Despite its relatively high success rate in FED, penetrating keratoplasty is not risk-free. Postoperative astigmatism, suture-related problems, and ineffective wound healing remain as significant issues that prolong the visual recovery period (119,120). Recently, several innovative surgical techniques for selective endothelial transplantation have been described with promising early results (121, 122, 123, 124, 125, 126, 127, 128).
Selective transplantation of a disk of posterior stroma and endothelium through a large scleral incision (deep lamellar endothelial keratoplasty) minimizes surgically induced astigmatism, as the anterior surface of the cornea remains intact (123,127). However, this technically demanding approach currently requires manual lamellar dissection, therefore creating interface irregularities that reduce best-corrected postoperative vision. This technique also theoretically reduces the risk of wound dehiscence, as the wound is created in the vascularized sclera, rather than the avascular cornea.
In microkeratome-assisted endokeratoplasty (121,122), an anterior corneal flap is created mechanically. The flap is reflected in the operating room and a corneal button of posterior stroma and endothelium is transplanted, as in a full-thickness keratoplasty. Infant donor cornea, which has very high cell counts but lacks the tensile strength to support a full-thickness graft, has been used with this technique with good success (124). Because endothelial loss is a major factor in graft failure in FED patients, the ability to use infant tissue may represent a significant advance in the management of this disease.
Finally, successful in vitro transplantation of a thin scroll of Descemet’s membrane carrying viable endothelium through a small scleral incision has been described (126). These novel techniques are still in their infancy, but have the potential to revolutionize the surgical management of Fuchs’ dystrophy.
Although corneal endothelial cells do not divide in vivo, they can be stimulated to proliferate in culture (8,129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139). Their growth can be modulated by diverse factors including fibroblast growth factor, epidermal growth factor, and endothelial cell growth factor (129,130,132, 133, 134,138,139). The recent demonstration that cultured human corneal endothelial cells can form a high cell density monolayer following ex vivo transplantation to recipient human cornea provides a foundation for the future advancement of endothelial replacement techniques (8), especially in combination with the surgical techniques described above. Future work may enable us to enhance the cell density of donor tissue prior to surgery. Ultimately, it may be possible to stimulate in situ repair and controlled proliferation of the corneal endothelial cells with exogenous growth factors or with gene therapy.
CONGENITAL HEREDITARY ENDOTHELIAL DYSTROPHY
Congenital hereditary endothelial dystrophy (CHED) was first described in 1863 as “corneitis interstitialis in utero” (140). It was initially classified as a variant of intrauterine interstitial keratitis (140), and subsequently as a stromal dystrophy (141, 142, 143, 144). In 1960, Maumenee (145) was the first to hypothesize that CHED resulted from a primary dysfunction of the corneal endothelium, which was later confirmed by histopathologic and electron microscopy data (146, 147, 148, 149). In 1971, the name congenital hereditary endothelial dystrophy was suggested (149).
CHED can be inherited in either an autosomal-dominant (AD) or an autosomal-recessive (AR) pattern (150,151). Linkage analysis had localized the dominant form (CHED1) to chromosome 20, near the locus for PPMD (152,153). Recessive CHED (CHED2) is not linked to this region, suggesting that it is a genetically distinct entity (154, 155, 156).
Clinical Features
Congenital hereditary endothelial dystrophy is characterized by diffuse, noninflammatory corneal opacity with edema (Fig. 47-8). The disease is bilateral and tends to be symmetric. Marked impairment of vision is characteristic. CHED typically presents at birth or in early infancy, and is a common cause of childhood corneal opacification (39,157). The two forms of CHED have different clinical characteristics (157).
Children with dominant CHED (CHED1) have clear corneas at birth. Corneal clouding is first noted during the first or second year of life and slowly progresses over 5 to 10 years. Photophobia and epiphora are common and may be the presenting signs of the disease. As corneal opacification increases, however, these signs may actually decrease. Nystagmus is uncommon in CHED1. Vision tends to be better (in the range of 20/40 to 20/400) than in recessive CHED (39,157). Some authors have suggested that CHED1 is more appropriately termed infantile hereditary endothelial dystrophy, given its clinical characteristics (157).
In contrast, corneal clouding is present at birth or within the neonatal period in patients with recessive CHED (CHED2). Corneal opacification is dense at the time of diagnosis, and does not tend to progress. There is no associated photophobia or epiphora. Nystagmus is invariably present, presumably the result of severe corneal opacification at an early age (39,157).
The clinical findings in CHED include corneal opacification, which extends to the limbus without an intervening clear zone. The opacification can vary in severity from mild haze to a milky, ground-glass appearance. Extensive bilateral corneal edema, with corneal thickness up to three times normal, is common. Fine epithelial edema creates a
roughened corneal surface and distorts the light reflex. Descemet’s membrane is thickened, but no guttae are present. Secondary changes such as band keratopathy, subepithelial fibrosis, and spheroidal degeneration may accompany the extensive edema, more commonly in the dominant variant (39,149,147,157). Subepithelial amyloid deposits resembling gelatinous droplike keratopathy have been reported in CHED2 (158,159). Painful epithelial erosion rarely occurs in CHED because bullous keratopathy is not characteristic, in contrast to Fuchs’ dystrophy.
roughened corneal surface and distorts the light reflex. Descemet’s membrane is thickened, but no guttae are present. Secondary changes such as band keratopathy, subepithelial fibrosis, and spheroidal degeneration may accompany the extensive edema, more commonly in the dominant variant (39,149,147,157). Subepithelial amyloid deposits resembling gelatinous droplike keratopathy have been reported in CHED2 (158,159). Painful epithelial erosion rarely occurs in CHED because bullous keratopathy is not characteristic, in contrast to Fuchs’ dystrophy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


