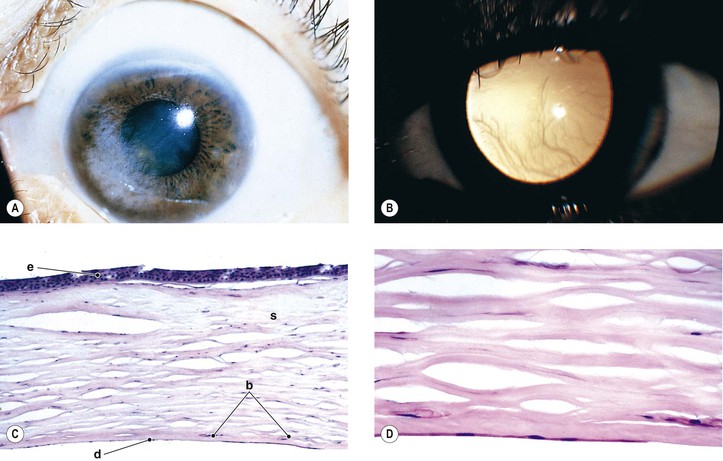
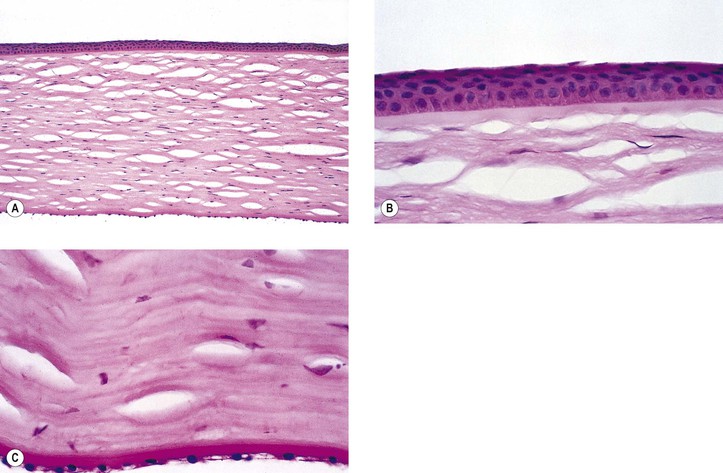
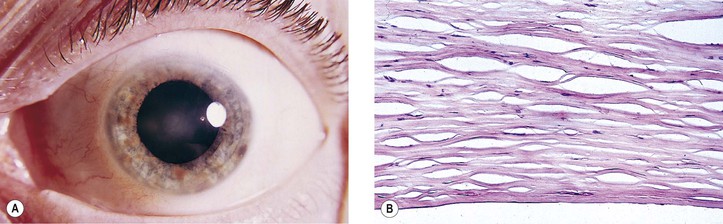
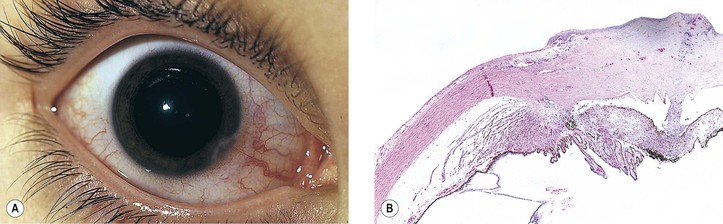
B. The cornea (Fig. 8.1) is a modified mucous membrane (it can also be considered, in part, as modified skin).
The cornea often is affected in association with cutaneous disorders; for example, low corneal sensitivity, abnormal tear quality, decreased cellular cohesion, squamous metaplasia of the conjunctiva, and goblet cell loss have been described in the Hallopeau–Siemens subtype of dystrophic epidermolysis bullosa. Specific corneal abnormalities in this disorder may include recurrent corneal erosion and superficial punctate corneal erosions.
Intermixed within the corneal epithelium are Langerhans’ cells (bone marrow-derived, CD Ia-expressing, dendritic-appearing cells) and occasional dendritic melanocytes. Langerhans’ cell histiocytosis has presented as a limbal nodule in an adult.
Three major types of molecules are found in the basement membrane: type IV collagen, heparan sulfate proteoglycans, and noncollagenous proteins (e.g., laminin, nidogen, and osteonectin). The basement membrane represents an important physiologic barrier between the epithelium and the stroma.
In healing large corneal abrasions that reach the limbus, the stem cells regenerate new corneal epithelium by a process called conjunctival transdifferentiation. First, the healing epithelium shows conjunctiva-like appearance, even to containing goblet cells, but then, slowly, it is transformed into a more cornea-like appearance without goblet cells.
Abnormalities of corneal epithelium can be demonstrated clinically by the use of fluorescein or rose Bengal. Fluorescein staining is enhanced when disruption of cell–cell junctions occurs, whereas rose Bengal staining is seen with deficiency of protection by the preocular tear film.
Absence of the cornea is a very rare condition usually associated with absence of other parts of the eye derived from the primitive invaginating ectoderm (e.g., the lens).
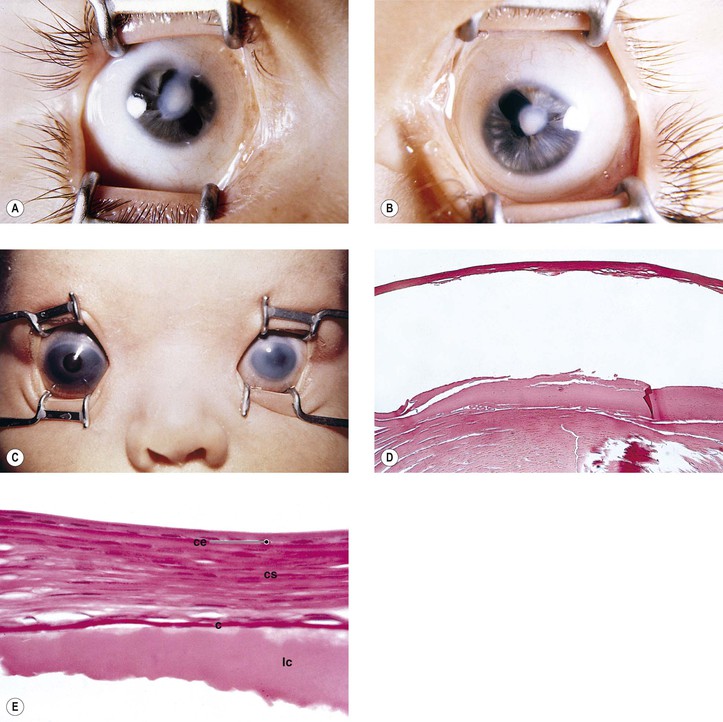
Microcornea may be associated with other ocular anomalies such as are found in microphthalmos with cyst, trisomy 13, and the Nance–Horan syndrome (X-linked disorder typified by microcornea, dense cataracts, anteverted and simplex pinnae, brachymetacarpalia, and numerous dental anomalies; there is provisional linkage to two DNA markers—DXS143 at Xp22.3–p22.2 and DXS43 at Xp22.2).
A lack of myofilaments and desmin in the cytoplasm of the anterior layer of iris pigment epithelium suggests that congenital microcornea may result from a defect of intermediate filaments.
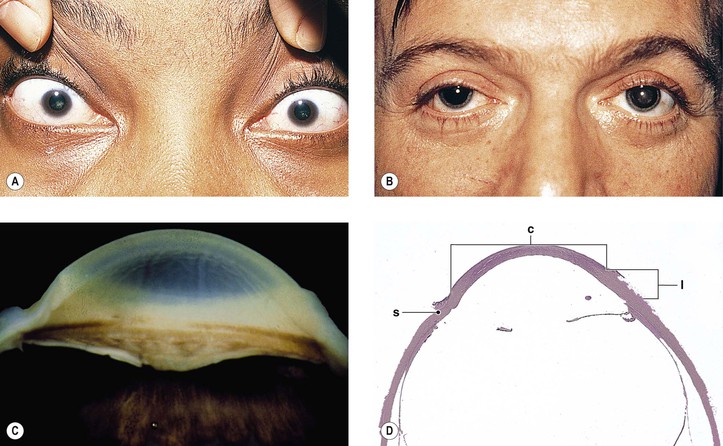
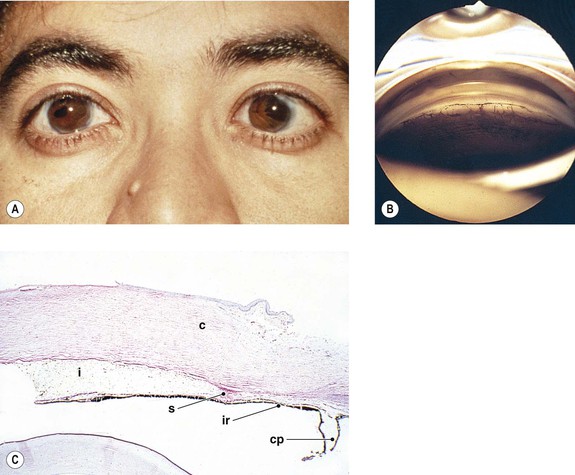
Cataract and subluxated lens commonly develop in adulthood. Glaucoma may result secondary to the subluxated lens. Rarely, megalocornea is associated with renal cell carcinoma.
Megalocornea, usually an isolated finding, may also be associated with ichthyosis, poikiloderma congenitale, Down’s syndrome, mental retardation, dwarfism, Marfan’s syndrome, craniostenosis, oxycephaly, progressive facial hemiatrophy, osteogenesis imperfecta, multiple skeletal abnormalities, nonketotic hyperglycemia, and tuberous sclerosis.
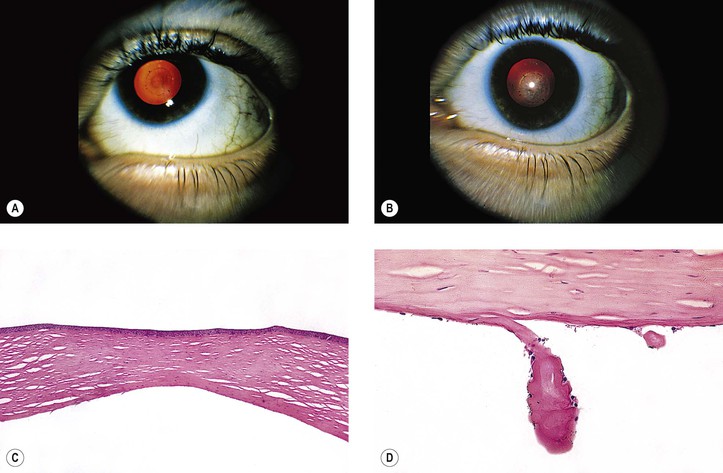
Mutations in keratocan are the probable mechanism of the cornea plana phenotype. A point mutation in exon 3 (937C>T), resulting in replacement of arginine by a stop codon at position 313 of keratocan protein, has been associated with autosomal-recessive cornea plana, variable anterior chamber depths, and short axial lengths.
The main theories of causation are arrested development during embryogenesis, intrauterine inflammation, and trauma.
In a study of 72 eyes of 47 patients with congenital corneal opacities, Peters’ anomaly was the most common cause (40.3%), followed by sclerocornea (18.1%), dermoid (15.3%), congenital glaucoma (6.9%), microphthalmia (4.2%), and birth trauma and metabolic disease (2.8%). 9.7% were idiopathic.
When iris is adherent to the posterior surface of the cornea beneath a region of corneal scarring, the resulting condition is called an adherent leukoma.
Peters’ anomaly may be part of a syndrome that includes microcephaly with cortical migration defects, and multiple intestinal atresias. It is believed to be a multiple vascular disruption syndrome.
Peters’ anomaly may be associated with deletion of short arm of chromosome 4 (Wolf–Hirschhorn syndrome), partial trisomy 5p, mosaic trisomy 9, deletion of long arm of chromosome 11, deletion of 18q, ring chromosome 21, interstitial deletion 2q14q21, translocation (2q;15q), ring 20 chromosomal abnormality, trisomy 13, the Walker–Warburg syndrome, and the fetal alcohol syndrome (see Fig. 2.14). Also, in a family that has Axenfeld syndrome and Peters’ anomaly, the condition was caused by a point mutation (Phe112Ser) in the FOXC1 gene.
Similar findings have been reported in cerebro-ocular myopathy syndrome.
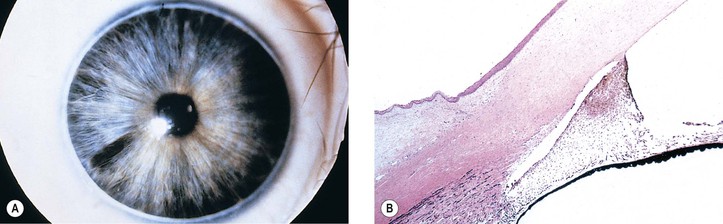
Peripheral dysgenesis of the cornea and iris includes a wide spectrum of developmental abnormalities, ranging from posterior embryotoxon (Axenfeld’s anomaly) to extensive anomalous development of the cornea, iris, and anterior chamber angle associated with systemic abnormalities (Rieger’s syndrome).3 An associated congenital glaucoma may occur, but the presence or absence of glaucoma does not necessarily depend on the degree of malformation. The abnormalities may be congenital, noninfectious, and noninherited (e.g., as part of trisomy 13 and partial trisomy 16q); congenital and inherited (e.g., Rieger’s syndrome); or congenital and infectious (e.g., rubella syndrome).
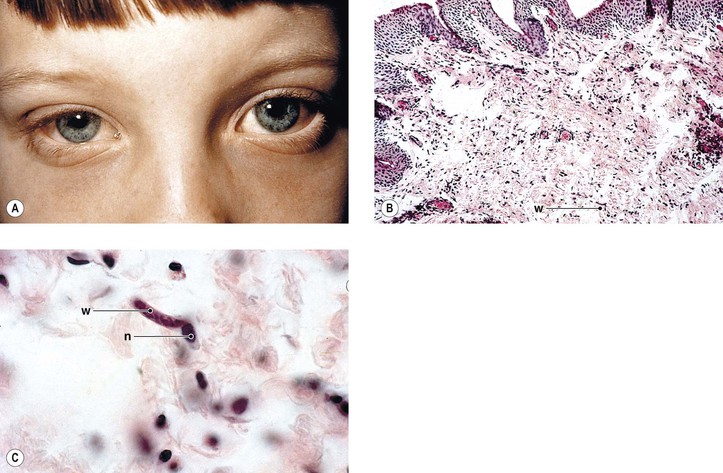
Axenfeld–Rieger anomaly has been linked to chromosome 6p25 (FKHL7 gene). Posterior embryotoxon (along with microcornea, mosaic iris stromal hypoplasia, regional peripapillary retinal depigmentation, congenital macular dystrophy, and anomalous optic discs) may be associated with arteriohepatic dysplasia (Alagille’s syndrome), an autosomal-dominant intrahepatic cholestatic syndrome. Posterior embryotoxon, iris abnormalities, and diffuse fundus hypopigmentation, together with neonatal jaundice, are highly characteristic of Alagille’s syndrome, which also has a strong association with optic drusen. Another association may be with oculocutaneous albinism.
Axenfeld–Rieger’s syndrome can be found as part of the SHORT syndrome (short stature, hyperextensibility of joints or hernia, ocular depression, Axenfeld–Rieger’s syndrome, and teething delay). Axenfeld–Rieger’s syndrome is different from iridogoniodysgenesis, which does not have a linkage to the 4q25 region (see later).
Genetic causes of Axenfeld–Rieger syndrome include mutations, deletions, or duplications of the forkhead-related transcription factor FOXC1, as mutations of the homeodomain (HD) protein PITX2 (PITX2 gene; chromosome 4q25). Axenfeld–Rieger syndrome caused by a deletion of the paired-box transcription factor PAX6 has been reported. Axenfeld syndrome and Peters’ anomaly caused by a point mutation (Phe112Ser) in the FOXC1 gene has been reported, as has familial anterior segment dysgenesis syndrome, which includes iris and corneal abnormalities and cataracts, showing random aggregates of small-diameter filaments that stain positively for cytokeratin.
Sclerocornea has been described in Mietens’ syndrome. It is also found in microphthalmia with linear skin defects syndrome, in which it has been present bilaterally. Sclerocornea is mainly a clinical descriptive term, and a distinct clinicopathologic entity of sclerocornea probably does not exist.
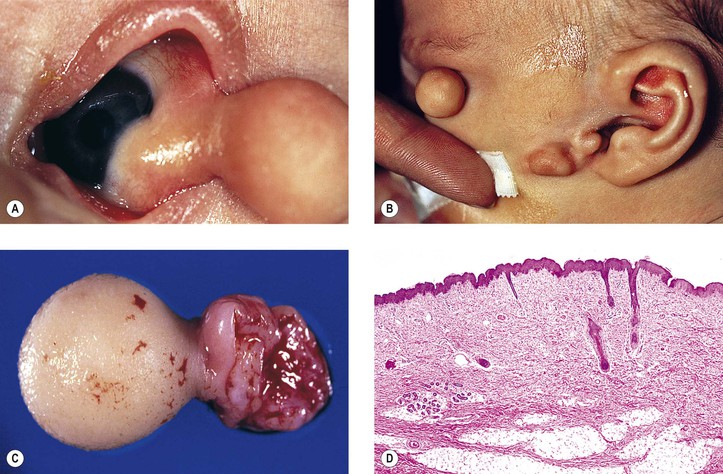
Goldenhar described the triad of epibulbar dermoids, auricular appendages, and pretragal fistulas in 1952. Eleven years later, Gorlin showed the added association with microtia and mandibular vertebral abnormalities (i.e., oculoauriculovertebral dysplasia).
Upper-eyelid colobomas commonly occur, but lower-eyelid pseudocolobomas are more often associated with the Treacher Collins–Franceschetti syndrome. Epibulbar choristoma, similar to that seen in Goldenhar’s syndrome, has been seen in association with nevus sebaceus of Jadassohn.
Encephalocraniocutaneous lipomatosis (congenital neurocutaneous syndrome including epibulbar choristomas and connective tissue nevi of the eyelids) should be considered, along with the sebaceous nevus and the Goldenhar–Gorlin syndromes, in the differential diagnosis of epibulbar choristomas.
Endothelial abnormalities have been reported in an African American family having a syndrome also characterized by abnormal craniofacial features and absence of the roof of the sella turcica. Other findings include abnormalities in the maintenance of retinal bipolar cells and of bipolar cells of the auditory system. One variation and one mutation of the homeobox transcription factor gene, VSX1 (RINX), characterize this family.
The evanescent subepithelial opacities, unlike the fine or medium-sized ones with adenoviruses types 3, 4, and 7, tend to be like a cluster of coarse, tiny bread crumbs. Similar findings occur with adenovirus type 19. Adenoviruses 3 and 7 are the most common causes of sporadic EKC. Other types (e.g., 1, 2, 4–6, 9–11, 13–15, and 29) also may cause moderate to severe EKC. Among the many other causes of subepithelial keratitis are rosacea, pharyngoconjunctival fever, onchocerciasis, and Crohn’s disease. The causes of nummular keratitis must also be considered [e.g., Dimmer’s (and related processes of Westhoff and of Langraulet) nummular keratitis and the similar interstitial keratitis of Epstein–Barr virus infection, inclusion conjunctivitis (Chlamydia), herpes simplex and herpes zoster infection, and brucellosis].
Uveitis and peripheral anterior and posterior synechiae commonly cause secondary angle-closure glaucoma. Chorioretinitis secondary to posterior involvement also occurs. The glaucoma and chorioretinitis, along with the keratitis, are common causes of the blindness. The ubiquitous bacteria, Wolbachia, colonizes the major pathogenic filarial nematode parasites of humans, including Onchocerca volvulus, and may contribute significantly to the inflammatory reaction within the eye.
The small black fly, Simulium species, ingests the microfilariae from an infected person and transmits them to the next human it bites. Immunologic cross-reactivity of a recombinant antigen of O. volvulus to a host ocular component of 44,000 M antigen suggests that intraocular presentation of the cross-reactive parasite antigen by microfilariae is essential for development of the ocular disease. Other filarial nematodes that may involve ocular structures include Loa loa and organisms that cause filariasis (e.g., Wuchereria bancrofti and Brugia malayi).