Connective Tissue/Collagen Vascular Diseases
C. Stephen Foster
The acquired connective tissue and vasculitic disorders, including rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, reactive arthritis, systemic lupus erythematosus (SLE), polyarteritis nodosa (PAN), Wegener’s granulomatosis, giant cell arteritis (GCA), relapsing polychondritis, juvenile idiopathic arthritis (JIA), scleroderma, polymyositis, and Sjögren’s syndrome may all produce conjunctival, episcleral, sclera, corneal, and intraocular inflammation. Additionally, except in the case of rheumatoid arthritis, the ocular manifestations of the disease may be the feature that stimulates the patient to first seek medical care. Furthermore, for reasons that are not well understood, the eye may be one of the most exquisitely sensitive indications of very ominous, otherwise subclinical disease activity. That is, ocular inflammation may portend catastrophic extraocular vasculitis in a patient whose systemic disease appears to be in remission. Examples of this are abundant and legendary, yet few ophthalmologists and even fewer rheumatologists are aware of this phenomenon. But the evidence for this is vast (1, 2, 3, 4, 5). Failure to act on the occurrence of new-onset ocular inflammation in such patients with dramatically increased therapeutic vigor is generally associated with disastrous systemic outcomes. This will be addressed more fully (below) for each specific disease. But this introductory preamble is intended to indicate to the reader the primary importance of this chapter: recognition of not only the ocular manifestations of the acquired connective tissue and vasculitic disorders, but also the systemic prognostic significance of same.
RHEUMATOID ARTHRITIS
Rheumatoid arthritis (RA) is the preeminent autoimmune disease, with (especially) genetically predisposed (6, 7, 8) individuals triggered (probably) by as yet unknown microbial contact to mount inappropriate immune, inflammatory attacks on various targets, including molecules in joint synovium, with resultant chronic inflammation that produces damage to the affected joints (9, 10, 11, 12, 13). The arthritis is generalized and is generally symmetric, and any joint can be involved (14, 15, 16).
Systemic Manifestations
Epidemiology
The prevalence of RA is 0.5% to 1.0% in most population groups, with higher rates in some Native American groups and lower rates in Asians and Africans. Women are affected three times more frequently than men.
Pathogenesis
The evidence for a combined microbiologic, immunologic, and genetic etiology for RA is compelling (16,17). Figure 23-1 illustrates this general paradigm.
Rheumatoid factor, the classic autoantibody produced in RA, is present in the circulation and in the joint, and is present in immune complexes, which are central to the cascade of inflammatory events (18), resulting in the release of a panoply of inflammatory cytokines and destructive enzymes that result in arthritis, scleritis or tissue destruction, and scarring (9,11,13,16,19, 20, 21). The articular manifestations of RA may be ascribed to the production of a number of cytokines, including interleukin-1 (IL-1), IL-2, IL-6, and IL-8; interferon-γ; macrophage-colony-stimulating factor; tumor necrosis factor-α; and epidermal growth factor. Many of the extraarticular manifestations of RA may also be attributed to systemic effects of circulating cytokines (22).
The important role of immune complexes is suggested by findings of increased levels of immune complexes and decreased complement levels (complement consumption in the process of immune complex formation) in synovial, pericardial, and pleural fluids of RA patients (18,23, 24, 25, 26, 27, 28, 29). Cell-mediated immunity (CMI) is also important in the pathogenesis of RA. T lymphocytes predominate in the synovial infiltrate (18,23, 24, 25, 26,28,29), with CD4+/CD29+ memory T cells prominent (22). Macrophages isolated from the synovium of patients with RA are capable of
inducing neovascularization, probably via secretion of IL-1, and therefore contribute to the development of the rheumatoid synovial pannus (30).
inducing neovascularization, probably via secretion of IL-1, and therefore contribute to the development of the rheumatoid synovial pannus (30).
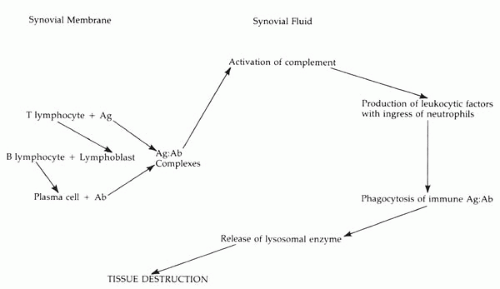 FIGURE 23-1. Proposed pathogenesis of rheumatoid arthritis. Ab, antibody; Ag, antigen. (From Zizic TM, Stevens MB. Rheumatoid arthritis and variants. In Ryan SJ Jr, Smith RE, eds. Selected topics on the eye in systemic disease. New York: Grune & Stratton, 1974:315-338, with permission.) |
Clinical Features
The average age of onset of RA is between 30 and 40 years. A prodrome of fatigue, malaise, loss of appetite, and weight loss may occur, with subsequent onset of morning stiffness, and then pain and swelling of the interphalangeal and metacarpophalangeal joints in the fingers and wrists. Larger joints, such as knees, ankles, shoulders, and elbows, become inflamed subsequently. The chronic progressive nature of the arthritis can lead to crippling joint damage (Fig. 23-2).
The course of RA varies from a short, mild episode of arthritis to progressive, severe, crippling disease. The longer the inflammatory activity, the less the likelihood of a remission. The sooner effective therapeutic intervention occurs, the less disability that ensues. This latter realization has prompted reevaluation of the traditional base-down pyramid model for RA therapy, with the most potent treatments (e.g., immunosuppressive medications) reserved for the apex of the pyramid, employed for only the most severe cases that had failed lesser therapy (nonsteroidal antiinflammatory agents, sulfonamides, gold). Evidence-based outcomes studies clearly indicate better outcomes, with less disability, when RA is treated more aggressively earlier in its course (31). A reappraisal of the role of chronic daily oral corticosteroid therapy has even arisen recently for this same reason (32).
Numerous extraarticular problems are part of the RA syndrome (33). Cardiac, respiratory, central nervous system, cutaneous, eye, and reticuloendothelial system damage may occur (Table 23-1). Pulmonary complications, including pleurisy with or without effusion, rheumatoid pneumoconiosis (Caplan’s syndrome), nonpneumoconiotic intrapulmonary rheumatoid nodules, and diffuse interstitial pulmonary fibrosis, are more common in men (34). Peripheral neuropathy, with a mixed sensorimotor component, occurs in less than 5% of patients with advanced RA. This neuropathy is probably caused by vasculitis of the vasa vasorum of the peripheral nerves (16,33). Cutaneous manifestations of RA include the characteristic subcutaneous nodules that occur in about 25% of patients (16). Raynaud’s phenomenon, erythema nodosum, and ischemic necrosis secondary to vasculitis may also occur (35). Generalized lymphadenopathy and splenomegaly develops in some patients with RA. Felty’s syndrome is characterized by rheumatoid arthritis, splenomegaly, and neutropenia.
The most obvious pathologic features of RA occur in the involved joints and surrounding tissue (9,16,33,36, 37, 38). The lesions probably begin in the synovial membrane with a proliferation of cells lining the synovium, with inflammatory cell infiltration (lymphocytes, plasma cells, and macrophages), and eventual granuloma formation, with epithelioid cells and even, in some instances, multinucleated giant cells.
Exudation of fluid into the synovial space occurs as a result of the inflammatory reaction, and cartilage and periosteal involvement within the joint capsule ensues, resulting in erosion of bone, formation of granulation tissue, and ultimately, destruction of the joint itself, with replacement by fibrous tissue. Loss of structure and function of the joint results in the characteristic deformities of RA (Fig. 23-3).
Exudation of fluid into the synovial space occurs as a result of the inflammatory reaction, and cartilage and periosteal involvement within the joint capsule ensues, resulting in erosion of bone, formation of granulation tissue, and ultimately, destruction of the joint itself, with replacement by fibrous tissue. Loss of structure and function of the joint results in the characteristic deformities of RA (Fig. 23-3).
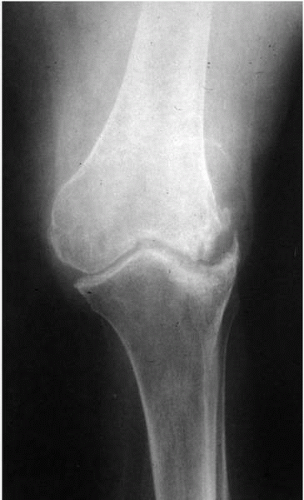 FIGURE 23-2. Typical radiographic pattern of a knee affected by rheumatoid arthritis, with narrowing of the joint space, along with new bone formation and obvious erosion on one of the tubercles. |
Ocular Manifestations
Potential ocular manifestations of RA include keratoconjunctivitis sicca (Sjögren’s syndrome), scleritis, episcleritis, peripheral ulcerative keratitis (PUK), sclerosing keratitis, and keratolysis (23,40, 41, 42). Ocular complications may occur in patients with limited systemic disease but are more common in those with marked systemic complications including vasculitis, pericarditis, and rheumatoid nodules.
TABLE 23-1. EXTRAARTICULAR MANIFESTATIONS OF RHEUMATOID ARTHRITIS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Keratoconjunctivitis Sicca
The most common ocular manifestation of RA is keratoconjunctivitis sicca (41,43). Sjögren’s syndrome (dry eye and dry mouth) is present in some patients. The dry eye associated with RA is indistinguishable from dry eye in the nonrheumatoid patient. However, unexplained xerostomia and RA (or other collagen vascular diseases) with or without swelling of the salivary glands complete the syndrome described by Sjögren (23). The presence of human leukocyte antigens
HLA-DR3 (44), HLA-B44, and HLA-DR4 (45), hypergammaglobulinemia, and reduced T lymphocytes are frequent findings in patients with RA and Sjögren’s syndrome (6,40), and the haplotypes serve to distinguish patients with these ailments from those without RA. Tear lysozyme is deficient or absent in Sjögren’s syndrome (40). Increased amounts of immunoglobulin A (IgA)-, IgM-, and IgG-containing immune complexes are present in patients with primary Sjögren’s syndrome and in RA patients, and IgA-containing immune complexes are more frequently found in Sjögren’s patients with extraglandular manifestations (46).
HLA-DR3 (44), HLA-B44, and HLA-DR4 (45), hypergammaglobulinemia, and reduced T lymphocytes are frequent findings in patients with RA and Sjögren’s syndrome (6,40), and the haplotypes serve to distinguish patients with these ailments from those without RA. Tear lysozyme is deficient or absent in Sjögren’s syndrome (40). Increased amounts of immunoglobulin A (IgA)-, IgM-, and IgG-containing immune complexes are present in patients with primary Sjögren’s syndrome and in RA patients, and IgA-containing immune complexes are more frequently found in Sjögren’s patients with extraglandular manifestations (46).
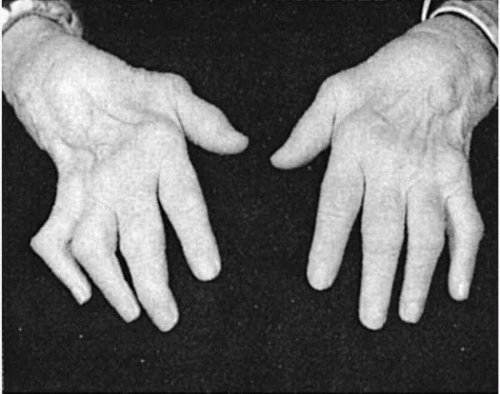 FIGURE 23-3. Characteristic joint deformities in the hands of a patient with rheumatoid arthritis. |
Clinical manifestations of keratoconjunctivitis sicca associated with RA are those of any dry eye, including foreign-body sensation, mucous threads in the tears, and the feeling of dryness and tightness around the eyes, often worse later in the day as tears evaporate (41). Examination discloses a reduced marginal tear strip, rose Bengal staining of the conjunctiva in the interpalpebral area, corneal complications related to dry eye (superficial punctate epitheliopathy, filaments), and shortened breakup time of the tear film.
Scleritis and Episcleritis
Inflammation of the collagenous coats of the eye (sclera and cornea) is a serious complication of RA (23,39,47, 48, 49, 50, 51). Scleritis (Fig. 23-4) is the most common, although episcleritis and keratitis may occur. Episcleritis, nodular or diffuse, is a relatively benign problem that responds well to low-dose oral nonsteroidal antiinflammatory agents. Topical steroid therapy for episcleritis is also effective, but this treatment probably prolongs (through multiple recurrences) the total duration or “life” of the problem. Scleritis (diffuse, nodular, necrotizing (with or without {scleromalacia perforans} inflammation), and posterior) is always a serious matter, both ocularly and systemically (39,40,52, 53, 54, 55, 56, 57).
Patients with scleritis characteristically have pain. Patients with episcleritis do not. Scleritis pain may be intense, and may even awaken the patient from sleep. In nodular episcleritis, the nodule is movable over the underlying sclera. The nodule of nodular scleritis is tender to the touch and immobile. The conjunctival vascular dilation associated with episcleritis can be blanched by the use of topical phenylephrine hydrochloride (Neo-Synephrine) or epinephrine. After the overlying vessels are blanched, dilatation of the vessels in the episclera can be observed. As emphasized by Watson (56) and by Foster (58), the pattern of vessel dilatation (episcleral versus scleral) is helpful in differentiating episcleritis from scleritis.
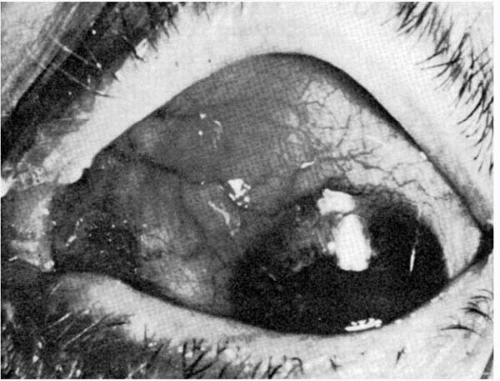 FIGURE 23-4. Localized scleritis in a patient with rheumatoid arthritis. |
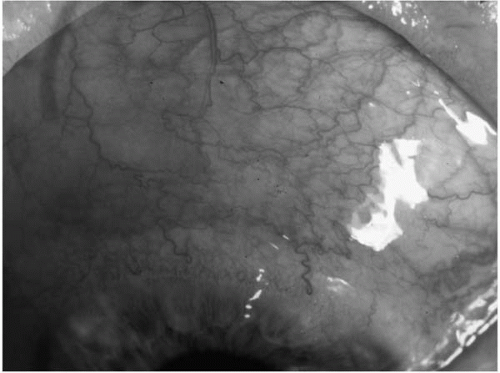 FIGURE 23-5. Scleritis, in a patient with a painful, tender eye. Note the violaceous hue to the ocular redness, as well as the loss of radiality of some of the episcleral vasculature. |
There is a blue hue to the red eye associated with scleritis, owing to the location of the inflammation and the associated vascular dilatation of the deeper layers of sclera (Fig. 23-5). The red eye of the patient with episcleritis lacks this blue hue and hence appears bright red (Fig. 23-6).
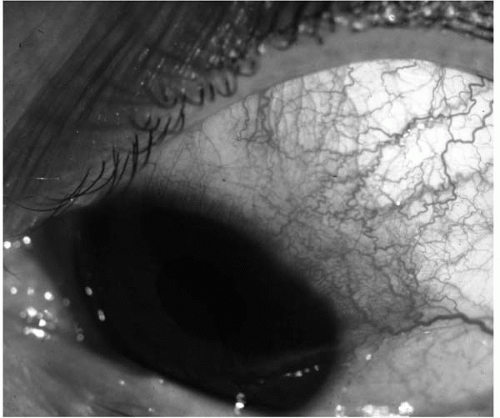 FIGURE 23-6. Episcleritis, in a patient with a red but nonpainful and nontender eye. Note the bright red color of the patch of sectorial episcleritis. |
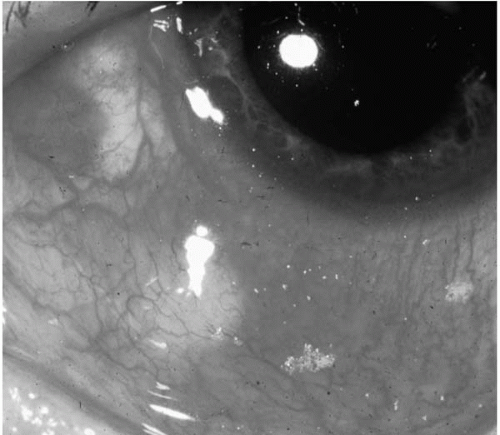 FIGURE 23-7. Nodular scleritis. Note the intensely red eye with a slight blue component to the redness. The obvious nodule is tender and immobile. |
Nodular scleritis is characterized by the presence of a painful, very tender, red elevated area of sclera inflammation. The nodule is immobile (Fig. 23-7).
Necrotizing scleritis is always associated with vasculitis and ischemia, with inflammatory destruction of sclera (Fig. 23-8). Scleromalacia perforans is an equally severe necrotizing and ulcerative process, but it is relatively asymptomatic in a deceptively quiet-appearing painless eye (Fig. 23-9). There is no clinically apparent inflammation. Slow, progressive ulceration of the sclera with exposure of underlying uvea occurs, however. This form of scleritis is always associated with long-standing, severe RA (39). The inflammatory and painful variety of necrotizing scleritis may be seen in RA as well, but it is also associated with other connective tissue disorders such as Wegener’s granulomatosis, polyarteritis nodosa, and relapsing polychondritis (23,39,59).
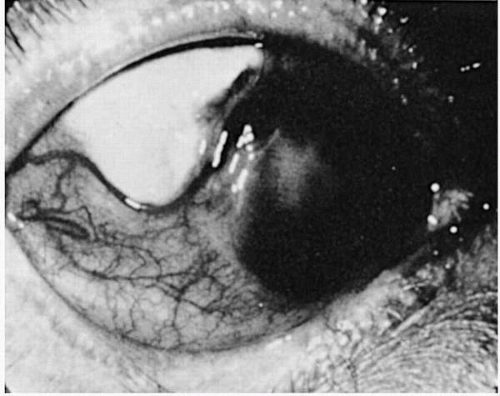 FIGURE 23-8. Active necrotizing scleritis and associated corneal marginal thinning and vascularization. |
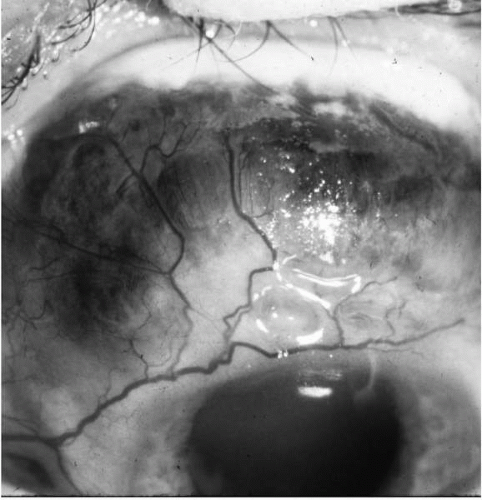 FIGURE 23-9. Scleromalacia perforans in a patient with rheumatoid arthritis. Note the lack of any clinically obvious inflammation (redness). The globe is not tender. |
Anterior segment fluorescein and indocyanine green angiography reveals vaso-obliterative vasculitis in all cases of necrotizing scleritis (60,61).
Posterior scleritis in the absence of anterior scleritis may be confused with retrobulbar problems (62). Pain accentuated with eye movement is present in almost all cases. Mild proptosis, diplopia, decreased vision, and limited ductions also occur. Exudative retinal detachment, posterior choroidal folds, and vitreitis may be present (62) (Fig. 23-10). Glaucoma may also occur, as a result of associated iridocyclitis or peripheral anterior synechia, or may be steroid-induced (63).
Ultrasonography is especially helpful in differentiating posterior scleritis from other entities (62,64) (Fig. 23-11).
Ultrasonography is especially helpful in differentiating posterior scleritis from other entities (62,64) (Fig. 23-11).
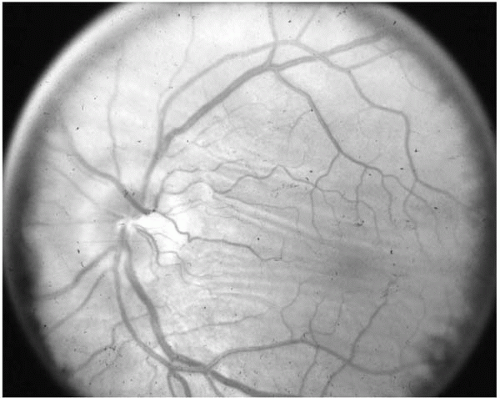 FIGURE 23-10. Fundus photograph of a patient with posterior scleritis. Note the retinochoroidal folds extending between the disk and the macula. |
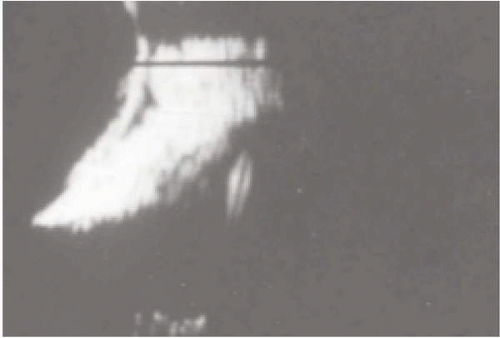 FIGURE 23-11. B-scan ultrasonography of a patient with posterior scleritis in the absence of anterior scleritis. The patient has pain, which is accentuated on binocular rotations. Note the thickening of the retinochoroid layer and the presence of edema in Tenon’s space, which, at the level of the optic nerve, produces the classic so-called T sign. |
Although the exact pathogenesis of scleral and episcleral inflammation in RA has not been established, it probably has the same cause as the joint inflammation (39,56). Immune complexes are probably deposited in the sclera of patients with rheumatoid scleritis, just as they occur in the synovia of joints in RA patients (20,40). These antigen-antibody complexes, with complement, trigger the cascade of events leading to scleral (or joint) inflammation (i.e., attraction of neutrophils and release of various enzymes and other mediators followed by localized tissue destruction) (39). Intrascleral vasculitis (microangiopathy) may also occur (65,66). This may result in deep scleral intravascular coagulation, ischemic necrosis, and scleral dissolution (39,65). Both scleral inflammation and vasculitis are probably present in some patients. Histopathologic studies of patients with severe scleritis have revealed the presence of active stromal fibrocytes in the absence of inflammatory cells, suggesting that collagenous resorption may precede granuloma formation. Mast cells were also abundant (67,68).
The pathologic changes of rheumatoid scleritis include foci of fibrinoid necrosis of the sclera surrounded by palisading fibroblasts and an inflammatory component consisting largely of neutrophils, lymphocytes, and plasma cells (36,63,69,70). The amount of cellular proliferation and necrosis varies, depending on the severity and the extent of vascular involvement.
Scleritis and episcleritis occur in approximately 4% to 10% of all RA patients (39,56). However, when a patient comes to an ophthalmologist with scleritis, RA must be considered, because it is the most common cause of scleritis (and episcleritis). The frequency of RA in patients presenting with scleritis is about 30% (12). Besides history taking for RA and other collagen disorders that may also be associated with scleritis, certain laboratory tests are appropriate. Rheumatoid factors (IgG, IgA, and IgM), uric acid, sedimentation rate, C-reactive protein, antineutrophil cytoplasmic antibody, fluorescent treponemal antibody absorption (FTA-ABS), purified protein derivative (PPD) (tuberculosis), and chest x-ray films (tuberculosis) are suggested screening tests. I also search for circulating immune complexes by the C1q binding and the Raji cell assays.
Keratitis and Sclerosing Keratitis
Corneal changes may be found adjacent to areas of scleral inflammation or as the only ocular complications of RA (23,39,56). Localized inflammatory cell infiltration of the cornea, followed by development of epithelial defects and frank loss of stroma, may occur (Fig. 23-12). A relatively severe proliferation and infiltration of inflammatory cells with secondary fibrovascular invasion results in peripheral corneal scarring in some cases.
Corneal involvement in RA includes keratitis and sclerosing keratitis, peripheral corneal ulceration (furrows), and keratolysis (39,56).
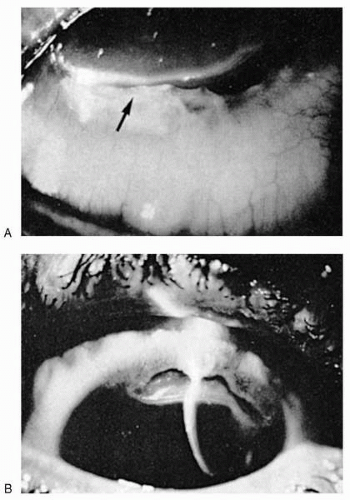 FIGURE 23-12. A: Limbal furrow in rheumatoid arthritis. There is no associated scleritis. B: Same patient as in A. Note the infiltrate (arrow) at the advancing edge of the furrow. |
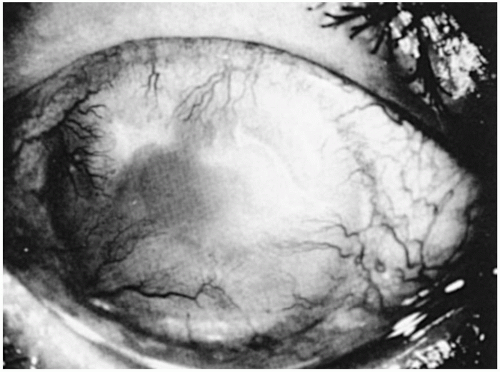 FIGURE 23-13. Sclerosing keratitis and keratolysis in a rheumatoid arthritis patient with scleritis. Note the marked vascularization of the cornea with thinning. |
Sclerosing keratitis is the most common corneal complication of scleritis (39,56). The keratitis occurs in association with severe scleritis and not as an isolated corneal finding (56). This is in contradistinction to peripheral guttering of the cornea, which may occur without scleral inflammation in patients with long-standing RA.
In sclerosing keratitis, a gradual gray corneal stromal thickening progresses toward the center of the cornea, followed by vascularization (Fig. 23-13). The advancing edge may have a striate configuration with crystalline-like changes developing behind this edge. If this process extends into the visual axis, it causes a marked decrease in vision. If the associated scleritis is nodular, sclerosing keratitis is generally localized to the quadrant of the scleral nodule. However, if the scleritis is diffuse, the entire cornea may become white and vascularized. Sclerosing keratitis may lead to perforation as a result of (noninfectious) localized corneal ulceration. Lipid deposition is a late change.
Keratitis alone (sterile corneal inflammation) may also be associated with scleritis and may occur in areas adjacent to the active scleral inflammation. Such corneal inflammation has been found in 30% to 70% of patients with scleritis or episcleritis associated with RA (52,70). The corneal inflammation includes midstromal and superficial stromal infiltrates, depending on the nature of the scleritis. The infiltrates may become very diffuse and even result in corneal epithelial breakdown and ulceration. This corneal complication may take a sclerosing configuration, with ingrowth of vessels and connective tissue resulting in peripheral corneal scarring as noted above.
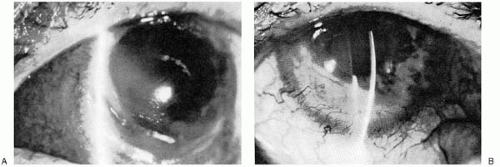 FIGURE 23-14. A: Marginal furrow with scleritis in rheumatoid arthritis. B: Same patient as in A, after local corticosteroid therapy. Note the decreased corneal edema and infiltrate and the increased neovascularization. |
Peripheral Corneal Ulceration (Furrows)
Peripheral corneal ulceration occurs with or without associated scleral or episcleral inflammation (Fig. 23-14) Brown and Grayson (72) reported marginal furrows in patients with chronic RA; this is probably a more common occurrence in RA patients than has been previously reported (23,73). The thinned area, usually located inferiorly, may be very superficial at first but progresses rapidly. The peripheral corneal ulceration is often associated with a mildly inflamed conjunctiva. This mild contiguous conjunctivitis should not be confused with episcleritis.
There is usually no vascularization in the bed of the furrow, and the corneal epithelium is intact. Limbal ulceration (52,57,73,74) may also occur in association with rheumatoid scleritis, episcleritis, or sclerosing keratitis. Although the majority of such ulcers do not progress, some have resulted in marked thinning and descemetocele formation. Perforation is rare but may occur with minor trauma.
The pathogenesis of the corneal lesions is unclear. Collagenase has been found in the conjunctiva and corneal epithelium adjacent to the furrow itself (75). The pathogenesis of the scleral and corneal inflammation may be the same as that for joint disease (40); cells associated with synovial tissue (and by analogy, with scleral or corneal collagen) produce an IgG antibody in response to an unknown antigenic
stimulus, possibly a virus, the IgG antibody may be altered, leading to the development of autoantibodies, immune complexes are formed with the abnormal IgG, and these are deposited locally in the synovium (or sclera) (40,74); these complexes activate the complement cascade, which triggers release of mediators of inflammation, attracts neutrophils, and causes tissue destruction and tissue melting. This sequence probably occurs in the sclera and the limbus as well as in synovium. Destruction of collagen and occlusion of associated vessels results in the scleritis, ischemic necrosis, and ulceration associated with rheumatoid syndromes.
stimulus, possibly a virus, the IgG antibody may be altered, leading to the development of autoantibodies, immune complexes are formed with the abnormal IgG, and these are deposited locally in the synovium (or sclera) (40,74); these complexes activate the complement cascade, which triggers release of mediators of inflammation, attracts neutrophils, and causes tissue destruction and tissue melting. This sequence probably occurs in the sclera and the limbus as well as in synovium. Destruction of collagen and occlusion of associated vessels results in the scleritis, ischemic necrosis, and ulceration associated with rheumatoid syndromes.
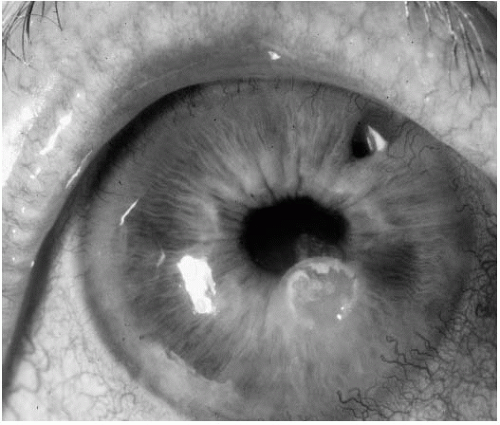 FIGURE 23-15. Paracentral keratolysis (stromal melting) with associated epithelial defect in a patient with rheumatoid arthritis. |
Paracentral Keratolysis
Ulceration of clear central or paracentral cornea may occur in eyes with no signs of inflammation (76) or less commonly, in cases of necrotizing scleritis (36,56,57) (Fig. 23-15). In most cases, severe aqueous tear deficiency is associated. The epithelium breaks down, superficial layers of the cornea begin to ulcerate, and a descemetocele may occur in the most severe cases. Perforation of the cornea may result rapidly, with or without minor trauma. Treatment options include the aggressive use of wetting agents, bandage contact lenses, application of tissue adhesive (prior to or after perforation), use of a protective shield, tarsorrhaphy, and tectonic keratoplasty (76, 77, 78). Patients with this problem usually respond poorly to these measures and often develop recurrent keratolysis.
Corneal Ulceration Following Cataract Surgery
A particularly devastating complication of cataract surgery in RA patients with keratoconjunctivitis sicca has been emphasized in recent years. Sterile corneal ulceration and melting have been observed days to weeks after otherwise unremarkable cataract surgery (39,79, 80, 81, 82, 83). Studies of penetrating keratoplasty corneal buttons has disclosed neutrophils localized near the areas of keratolysis, suggesting that collagenase derived from these neutrophils is involved in the pathogenesis of keratolysis (82). Patients who develop this problem are usually elderly women with moderate dry eyes and Sjögren’s syndrome, RA, or another collagen vascular disease. Physician failure to recognize the dry eye is a major problem. Prevention of epithelial damage at the time of cataract surgery and prompt institution of therapy for dry eyes or sterile corneal ulceration are critical. Unless the underlying cause is recognized, central corneal ulceration may progress to descemetocele formation, perforation, and loss of the eye.
Diagnosis and Differential Diagnosis
The diagnosis of RA is clinical. Arthritis, rheumatoid nodules, presence of rheumatoid factor, and diagnostic radiologic findings (84,85) form the basis for the diagnosis. Guidelines for the diagnosis of RA have been established by the American Rheumatism Association (15) and more recently, by the American College of Rheumatology (86).
The most characteristic laboratory feature of RA is the presence of rheumatoid factor (16,87). There are three rheumatoid factors, antibodies to IgG; classical rheumatoid factor is IgM anti-IgG; IgG anti-IgG and IgA anti-IgG may also be produced. One or more rheumatoid factors is present in about 90% of patients with RA. Numerous rheumatoid factor tests have been developed. The most commonly used tests include the Waaler-Rose test and the latex bead agglutination test. The first employs sheep red blood cells sensitized with rabbit antibodies to sheep cells. Rheumatoid factor combines with the rabbit IgG, causing agglutination of the sheep red blood cells. Test results are positive in 60% to 70% of patients with RA (16). The latex test involves adsorbing aggregated human 7S IgG on latex particles; rheumatoid factor reacting with IgG causes flocculation of the latex particles. Results are positive in 80% to 90% of patients. An increased frequency of positive test results, and higher titers occur in patients with subcutaneous nodules, in patients with RA variants (Felty’s, Sjögren’s), and in patients with multiple extraarticular manifestations of RA. The development of enzyme-linked immunosorbent assays for rheumatoid factor may increase the sensitivity of rheumatoid factor measurements (88).
Rheumatoid factor is present in some patients with connective tissue diseases other than RA (16), in up to 20% of patients with scleroderma, polyarteritis nodosa, and SLE. It is usually not present in patients with ankylosing spondylitis, enteropathic arthritis, reactive arthritis, or juvenile idiopathic arthritis. Therefore, testing for rheumatoid factor is helpful in differentiating variants of rheumatoid disorders.
Anemia, leukocytosis, and elevation of the erythrocyte sedimentation rate (ESR) and of the C reactive protein (CRP) are the other typically abnormal laboratory parameters in patients with RA. Additionally, approximately 25% of patients with classic RA are antinuclear antibody positive. If neutropenia is present, Felty’s syndrome should be suspected.
Treatment
Treatment of RA and its variants is very difficult and time-consuming and ideally should be orchestrated by a rheumatologist. Additionally, patients with RA have chronic problems that require not only medical treatment, but also rehabilitation services and even surgery to relieve deforming and incapacitating deformities in joints.
Almost 25% of RA patients respond initially to aspirin alone (16). Although there are potentially serious side effects associated with chronic salicylate use, salicylates remain a useful drug in the care of patients with RA. Salicylates inhibit prostaglandin synthesis. Therapeutic salicylate serum levels are 20 to 25 mg/dL. Enteric-coated aspirin may be required in patients who have gastric intolerance to the 2 to 4 g of aspirin per day generally required to achieve this serum level. If there is no clinical response despite adequate serum levels within 8 weeks, other nonsteroidal antiinflammatory drugs (NSAIDs) are generally prescribed. Naproxen, tolmetin sodium, ibuprofen, indomethacin, diclofenac, piroxicam, or one of the cyclooxygenase-2 specific (Cox-2 specific) NSAIDs may be effective. Gold salts (72,89, 90, 91, 92) and antimalarial drugs are also employed. Antimalarials that are quinoline derivatives have a number of potential side effects, including some related to the eye. The ophthalmologist must be aware of the corneal deposits and retinal toxic changes that may occur in the retinal pigment epithelium of patients taking certain quinoline derivatives. The positive effects of these drugs can generally be achieved at a lower dosage than that which usually results in ocular toxicity (93).
Systemic corticosteroids have a very potent antiinflammatory effect, relieve pain, and maintain function of the joints in patients with RA. Steroids work rapidly and can be used initially, whereas slower-acting remittable agents take effect. Low-dose corticosteroids are often used as a substitute for NSAIDs in patients who have gastrointestinal or other intolerance to NSAIDs (94), and a recent reappraisal of the safety and efficacy of chronic low-dose systemic steroid therapy for patients with RA suggests that low doses (≤10 mg/day) of prednisone used early (within 1 year of onset of the disease) has a disease modifying effect, limiting bony progression, with limited and manageable side effects [weight gain (3 kg), ecchymoses, and osteopenia); the drug should be stopped within 6 months. Pulsed high-dose intravenous corticosteroids (methylprednisolone) have also been used in patients with rapidly progressive RA, generally in conjunction with long-term steroid-sparing agents such as gold, penicillamine, or other immunosuppressive disease modifying agents (52,95, 96, 97). Immunosuppressive drugs, such as azathioprine and methotrexate, have a similar favorable effect on the inflammation. These medications used to be considered if other drugs failed to induce a remission (base-down pyramid model), because of the greater potential risks associated with the chronic use of immunosuppressive medications. The risk of increased susceptibility to cancer development after the use of cytotoxic agents is an especially important consideration in the management of RA patients, with particular concern about those who harbor the Epstein-Barr virus (98). Some studies have evaluated cyclosporine as a therapeutic agent in RA (99). Although these studies have generally demonstrated a significant improvement in disease activity, problems with nephrotoxicity have limited the usefulness of cyclosporine for care of patients with RA. Immunomodulatory agents are now being employed much earlier in the course of RA, because outcomes studies have shown, quite clearly, better outcomes employing this paradigm in place of the older, more conservative base-down pyramid one.
Biologics (monoclonal antibodies directed against specific cell types and against specific cytokines) have also revolutionized the care of patients with rheumatoid arthritis. Tumor necrosis factor-α (TNF-α) is a preeminently important inflammatory cytokine found in abundance in inflamed joint effusion fluid (unlike aqueous or vitreous humor of patients with uveitis, in which TNF-α is not impressive). Anti-TNF-α therapy [etanercept (Enbrel) was the first, but infliximab (Remicade) and adelimumab (Humira) have joined in the pack], usually in combination with methotrexate but sometimes even as monotherapy, is a major advance in our care of patients with arthritis, especially those with RA. IL-1 is also a keystone in the inflammatory pathways leading to joint destruction in uveitis, and antibody therapy directed against the IL-1 receptor [kineret (Anakinra)] is another “biologic” that has been approved by the United States Food and Drug Administration (FDA), based on safety and efficacy data, for the treatment of patients with rheumatoid arthritis.
Ocular Treatment
The management of keratoconjunctivitis sicca includes replacement tear therapy, occlusion of the lacrimal drainage puncta, warm compresses and lid massage (for the invariably present meibomian gland dysfunction), and, in selected cases, therapy aimed at any active inflammatory component of the disorder (topical cyclosporine and/or steroid).
The treatment of episcleritis and scleritis is directed toward the underlying disease. Topical steroids may help while systemic medication takes effect.
Episcleritis
Although highly effective, topical steroids actually prolong the total duration of recurrences of episcleritis. Therefore,
we advocate the avoidance of steroid therapy for patients with episcleritis, and instead use only iced artificial tears topically. An oral NSAID is used for those patients who must have prompt resolution of the episcleritis. NSAID therapy is also generally our first step in treating scleritis. I prefer the Cox-2 specific NSAIDs, celecoxib (200 mg b.i.d.) and rofecoxib (25 mg/day). Diclofenac (75 mg PO b.i.d.), sustained-release indomethacin (75 mg b.i.d.), diflunisal (500 mg b.i.d.), and naproxen (500 mg b.i.d.) are nonselective NSAID alternatives. Early response to therapy includes a dramatic decrease in the deep pain of scleritis. Systemic corticosteroids are employed (1 to 1.5 mg/kg/day of prednisone) in cases of scleritis that do not adequately respond to NSAID therapy.
we advocate the avoidance of steroid therapy for patients with episcleritis, and instead use only iced artificial tears topically. An oral NSAID is used for those patients who must have prompt resolution of the episcleritis. NSAID therapy is also generally our first step in treating scleritis. I prefer the Cox-2 specific NSAIDs, celecoxib (200 mg b.i.d.) and rofecoxib (25 mg/day). Diclofenac (75 mg PO b.i.d.), sustained-release indomethacin (75 mg b.i.d.), diflunisal (500 mg b.i.d.), and naproxen (500 mg b.i.d.) are nonselective NSAID alternatives. Early response to therapy includes a dramatic decrease in the deep pain of scleritis. Systemic corticosteroids are employed (1 to 1.5 mg/kg/day of prednisone) in cases of scleritis that do not adequately respond to NSAID therapy.
Immunomodulatory therapy is employed for scleritis resistant to all other forms of therapy and in cases associated with potentially lethal outcomes (necrotizing scleritis; scleromalacia perforans, cases associated with Wegener’s granulomatosis, polyarteritis nodosa, relapsing polychondritis). Cyclophosphamide, methotrexate, azathioprine, mycophenolate mofetil, cyclosporine, and chlorambucil have all been used in this context (19,100, 101, 102). Cyclophosphamide is the most effective agent (19,100,102). Hemorrhagic cystitis and severe bone marrow depression are the major potential complications of its use. I prescribe 2 to 3 mg/kg/day, and in urgent care administer the drug intravenously (1 g/m2). Guidelines for managing patients on this medication for eye disease have been published (19,100). Active involvement of an experienced chemotherapist (ocular immunologist or otherwise) is mandatory in the management of such patients, especially when systemic corticosteroids or immunosuppressives are employed. Clues to improvement include rapid relief of pain, decrease in redness, and improvement of vision.
We have considerable experience in the use of the various biologics (see above) in the care of both the ocular and extraocular manifestations of RA. Remicade has been especially effective in patients with scleritis. Another biologic, not mentioned above and not FDA approved for treatment of rheumatoid arthritis or for any eye disease, is Daclizumab (Zenapax), which has also been extremely valuable to us in our care of patients with otherwise treatment-resistant ocular inflammatory disease associated with RA. This monoclonal antibody is directed against the IL-2 receptor and hence blocks the binding of IL-2 to its receptor on T lymphocytes.
Management of severe thinning associated with scleritis is difficult. Most cases do not perforate spontaneously, although such eyes are more susceptible to rupture from trauma. Homologous scleral grafts can be very difficult to accomplish but may result in salvage of the eye (23). Autologous fascia lata or periosteum has also been successfully employed to reinforce or repair areas of severe scleral thinning or perforation (23,103, 104, 105, 106).
Therapy for sclerosing keratitis includes the use of high-dose topical corticosteroids to prevent progression of the corneal disease (56,65), and systemic therapy (corticosteroids, NSAIDs, and immunosuppressives), as outlined earlier for scleritis, is important to treat both the associated scleritis and the corneal disease.
Therapy for marginal or central thinning, ulceration, or melting associated with RA is controversial. It is important to exclude microbial infection in all cases. Keratoconjunctivitis sicca, microbial keratitis, lid margin and meibomian gland pathology, and scleritis must be identified and treated (23,107). If there is no infiltration in the corneal furrow, and a clear zone is present between the limbus and the ulcer, corticosteroids should not be used; the danger of perforation in this instance is too great (39,56,74). However, if the ulcer resembles a Mooren’s ulcer, beginning at the limbus, extending centrally and circumferentially, and having a major white blood cell infiltrative component, topical steroids may be helpful (23). Frequent examinations are essential, because perforation is a distinct risk, especially with the use of topical corticosteroids. Improvement and resolution of marginal ulcers may also occur with high-dose systemic corticosteroid therapy.
Topical collagenase inhibitors such as acetylcysteine (Mucomyst 20%), 1% medroxyprogesterone, and doxycycline have all been used in the treatment of corneal melting syndromes (8 times/day) (75). I especially like systemic doxycycline, 100 mg PO b.i.d., and topical medroxyprogesterone q2h for this purpose. Hydrophilic bandage lenses may be effective in promoting healing of the ocular surface. Tissue adhesive applied to the ulcer may also be helpful, as can resection of the conjunctiva adjacent to the marginal furrow.
Immunosuppressive therapy has been employed in unusually severe cases in which topical medication did not suffice (39,56,100,101). Azathioprine, cyclophosphamide, and chlorambucil have been used to treat not only severe rheumatoid systemic disease and severe scleritis, but also the associated corneal ulceration. Cyclophosphamide is probably the most effective. Dosages must be carefully monitored as noted earlier (108).
Perforation occurs in the corneal ring ulcer or furrow. If imminent, perforation can be prevented with a ring, patch, or lamellar corneal graft or fascia lata graft (39,106). The destructive process, however, will destroy this graft too, unless the underlying immunologically driven process is modified through systemic therapy (74,107).
JUVENILE IDIOPATHIC ARTHRITIS
There are several hundred thousand patients with JIA, the most important cause of chronic and progressive crippling childhood arthritis in the United States (109, 110, 111, 112). The prevalence of JIA is approximately 94 per 100,000 children, and the incidence is about 14 cases per 100,000 per year. The age of onset is generally between 2 and 4 years, with some cases occurring in the preteen years. JIA rarely occurs
before the age of 6 months. It occurs a little more frequently in girls than in boys. The iridocyclitis and associated complications are major causes of blindness in children in this country; therefore, recognition of this syndrome and accurate classification are important. JIA-associated iritis accounted for about 80% of all cases of uveitis in children in one series from England (109). Band keratopathy is the principal corneal feature of JIA (41,85,113).
before the age of 6 months. It occurs a little more frequently in girls than in boys. The iridocyclitis and associated complications are major causes of blindness in children in this country; therefore, recognition of this syndrome and accurate classification are important. JIA-associated iritis accounted for about 80% of all cases of uveitis in children in one series from England (109). Band keratopathy is the principal corneal feature of JIA (41,85,113).
There are three relatively distinct forms of JIA, classified on the basis of disease onset: systemic onset, polyarticular onset, and oligoarticular onset (109,110,114, 115, 116, 117).
Clinical Features
Systemic-onset JIA is a febrile, severe systemic illness with large joint involvement, splenomegaly, and lymphadenopathy (118). Iridocyclitis (or any other ocular finding) is rare in patients with systemic onset JIA (110,119). Polyarticular onset JIA accounts for about 50% of patients with JIA and is usually seen in younger children. Fever and lymphadenopathy may occur. Oligoarticular onset JIA begins with arthritis in five or fewer joints. Uveitis risk factors in JIA patients include female gender, oligoarticular onset, presence of circulating antinuclear antibodies, and the presence of HLA-DR5 or -DP2 (120). Children with oligoarticular-onset JIA are the most severely affected from the ocular standpoint (116,117). Although the arthritis may be very mild in such patients, the ocular complications are very severe and occur in up to 25% of patients (110,116,117,121). Chronic iridocyclitis is the predominant finding (122, 123, 124, 125, 126, 127, 128, 129). The presenting symptom may be decreased vision on a routine school examination or the onset of cataract with a white pupil observed by a parent or teacher. Most of the patients have no symptoms. By the time they are examined, they may have band keratopathy, cells and flare in the anterior chamber, posterior synechiae with a bound-down pupil, secondary cataract, and secondary glaucoma. The disease is usually present bilaterally. Because of the insidious onset and chronic nature of the ocular disease, all patients with JIA should be examined by an ophthalmologist every 3 months. In patients with iridocyclitis, close follow-up is important because increased inflammation is usually not accompanied by increased symptomatology (130).
The iridocyclitis associated with JIA is frequently relentlessly progressive despite corticosteroid therapy, with eventual blindness caused by cataract and glaucoma (131,132). Vitreitis, tractional retinal detachment, macular cysts and edema, macular holes, and lesions of the choroid are not rare (133). Pathologic findings are nonspecific, with infiltrates (mononuclear cells and lymphocytes) in the ciliary body and iris (124,134, 135, 136). Plasma cells have been prominent in the iris of some patients (137).
Patients with JIA and active iridocyclitis have been found to have elevated antibody levels to human iris and retina, compared with either normal control subjects or JIA patients without active iridocyclitis (138), and antibody to retinal S antigen is found more frequently in patients with JIA and uveitis, compared with normals and with JIA patients without uveitis (134).
Although the most important ocular complication of JIA is chronic iridocyclitis, corneal complications (band keratopathy) also produce visual morbidity. Band keratopathy frequently occurs in young patients with any form of chronic inflammation but is especially prominent in patients with JIA-associated uveitis (113). The keratopathy begins in Bowman’s layer, usually at the 9 and 3 o’clock position meridians, with deposition of calcium crystals. Involvement of the deep layers of the corneal epithelium progresses in a band-shape across the entire cornea. Holes in the calcium deposition may give it a Swiss-cheese appearance (Fig. 23-16). These holes may represent areas where nerves penetrate Bowman’s membrane, ending in corneal epithelium.
A number of entities are associated with band keratopathy. Although it occurs as a secondary change in patients with chronic uveitis, it can also occur in individuals without other major eye problems (41,140, 141, 142, 143, 144). In some cases, serum calcium levels may be elevated (145).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


