Clinicopathologic Correlates in Orbital Disease
Zeynel A. Karcioglu
Of all essential human senses, vision is the most important. The eye, organ of vision, should be well protected and supported functionally and structurally. This protection and support is provided by the orbit, which is a cone-shaped bony structure with a volume of 30 mL in which the 7-mL globe is positioned centrally and anteriorly. All the support systems of the globe, including the optic nerve, lacrimal gland, extraocular muscles, fibroadipose tissue, peripheral nerves, ganglionic tissue, and blood vessels are designed to be confined within approximately 25 mL of space surrounding the eyeball. Many tissues are crowded in this limited space and give origin to a variety of congenital, structural, inflammatory, degenerative, and neoplastic diseases. Further, orbital pathologies are not limited only to primary disorders; many systemic diseases and diseases of the globe may affect the orbit secondarily. The orbit is also secondarily affected by the diseases of the cranium, eyelids, conjunctiva, and the nasal cavity and paranasal sinuses. This chapter attempts to give an overview of the clinico-pathological correlation of orbital diseases. Because of limited space, entities that are encountered more often are detailed and some other less frequently seen pathologies are only mentioned with citations of pertinent references.
STRUCTURAL DISORDERS
CONGENITAL ANOMALIES
Orbitocraniofacial Deformities
The orbital bones begin to develop during the first 2 months of embryogenesis.1 Toward the end of the fifth week, the axes of the two orbits begin to move forward as a result of the growth of the maxillary processes.2 Most of the orbital bones are formed during the third month of gestation, but their complete ossification and fusing takes a longer time to complete, at approximately the seventh month of gestation. At term, the orbit is almost spherical in configuration. The periorbital sinuses begin to develop at approximately the second month of gestation and continue to grow after birth. Although the eye reaches adult size (approximately 7 mL) at approximately age of 2.5 years, the adult form and dimensions of the orbit (approximately 30 mL) are not finalized until puberty. Therefore, between the second month of embryogenesis and the age of puberty, numerous factors may affect the development of the orbit. For example, if the globe fails to develop or it is microphthalmic, the orbit does not attain its normal volume. However, if a congenital cyst or tumor develops within the orbit and expands its volume, the socket reaches a very large size unless the cyst is removed
Orbitocraniofacial deformities can be categorized in two large groups as craniosynostosis and clefting disorders.
Craniosynostosis
This developmental pathology of the orbit is dependent on the fusional abnormalities between the bones and it is divided into primary and secondary forms. Primary craniosynostosis refers to premature fusion of cranial sutures caused by embryologic error. Secondary synostosis is the premature closure of sutures because of other causes such as intrauterine trauma or effects of teratogenic drugs.3,4 Approximately 15% of craniosynostosis cases are associated with systemic abnormalities and are known as syndromic craniosynostosis; the remaining 85% represent nonsyndromic forms.5 Syndromic craniosynostosis represent a variety of inherited syndromes with abnormal development of the skull and orbit including: Apert syndrome with syndactyly of hands and feet, Crouzon syndrome with shallow orbits and proptosis, maxillary hypoplasia and cleft palates, and Pfeiffer syndrome with malformation of thumb and great toe and soft tissue syndactyly.6
Clefting Disorders
These malformations involve more extensive soft tissue abnormalities and depending on the location and the extent of the cleft, may be associated with bone malformations as well. The mapping system that is used to describe the location of a cleft is known as the Tessier clock face. In this scheme, the clefts are numbered from 0 to 14 beginning at the inferonasal area and moving clockwise to the superior nasal area. Commonly encountered clefting syndromes are: Goldenhar syndrome, Treacher-Collins syndrome, and the facial microsomias7,8 (Table 1).
TABLE 1. Summary of Orbito-Craniofacial Dysmorphic Syndromes | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
ANOPHTHALMOS/MICROPHTHALMOS
When the globe is abnormally developed, microphthalmos, congenital cystic eye, and extremely rarely, anophthalmos occur. Microphthalmos usually occurs as a unilateral condition and in approximately 10% of cases it is associated with other craniofacial malformations including agenesis of the corpus callosum, polymicrogyria, and mid-line arachnoidal cysts. Microphthalmos may be seen as a part of several genetically determined neuronal migration disorders such as Walker-Warburg syndrome, Aicardi syndrome, and Fukuyama congential muscular dystrophy.9,10
In cases of microphthalmos and anophthalmos the orbit may be well formed but does not develop to a full adult volume. The mechanism by which the presence of the globe effects the growth of the orbit is not well understood. Microphthalmos may be associated with a colobomatous cyst as a result of the abnormal closure of the embryonic optic fissure leading to the prolapse of neuroectodermal tissues into the orbit (Fig. 1) This cystic structure may increase rapidly in size to overshadow the abnormal globe and may be confused with a neoplasm. When cystic lesions in the orbit are suspected imaging studies should be performed not only to look for other intracranial abnormalities but also to establish the possible connection of the cyst to the colobomatous globe versus to abnormally formed meninges.11 Macrophthalmos (buphthalmos) may also rarely develop as a congenital anomaly in patients with Sturge-Weber syndrome and rarely in neurofibromatosis type I.
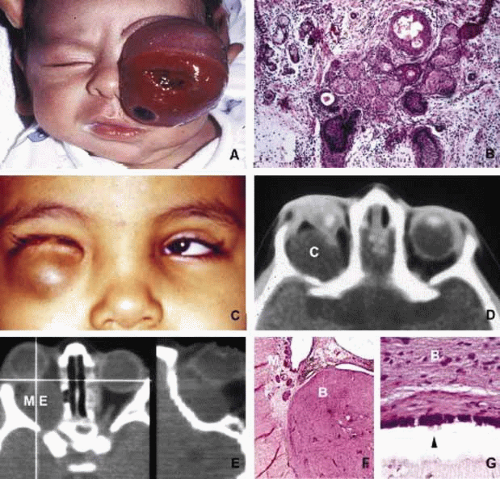 Fig. 1. Congenital lesions. A very large cystic teratoma of a 1-month old child (A). B. Histology of this lesion that contains a variety of tissues, endodermal, ectodermal, and mesenchymal. An orbital cyst (C) in an orbit containing micro-ophthalmic globe. C, D. The protrusion of the cyst inferiorly creates a mechanical lower lid ptosis that narrows the right maldeveloped conjunctival sac even further. Axial and sagittal CT scan showing a meningoencephalocele (ME) occupying the entire orbit (E). The histopathologic examination of the lesion revealed both meningeal (M) and brain (B) tissues (F). The high-power histopathology reveals ciliated ependymal cells lining some of the cystic spaces (arrowhead) (G). |
ORBITO-CRANIAL MALDEVELOPMENTS
Cephalocele results from the extension of maldeveloped CNS tissues including meninges (meningocele), brain parenchyma (encephalocele), and the combination of the two (meningoencephalocele) into the orbital cavity.12,13 Intraorbital cephaloceles may develop anteriorly at the suture lines of orbital bones or posteriorly extending into the orbit from orbital fissures and the optic canal. Depending on the combination of these herniations they contain brain and/or meningeal tissues (Fig. 1). Aberrant fibroglial tissue has also been described in the orbit.14
Hamartoma is a tumor-like proliferation of tissues that normally exist at a given body location. The best examples of orbital hamartomas are the vascular hamartomatous lesions that are composed of vascular elements including capillary endothelial cells, distended or collapsed cavernous blood and lymph vessels, tortuous arterial and venous channels with or without anastomoses, etc. Other examples of hamartomatous orbital tumors include neurofibroma and lipomatous hamartoma.15
Choristoma, however, is a tumor-like proliferation of tissues that are not normally present at a given body location. The most commonly encountered example of orbital choristoma is a dermoid.16 Dermoids that present with many varieties result from the entrapment of epithelial structures at the site of closure of fetal fissures. Superficial dermoid cysts occur primarily subcutaneously anterior to orbital septum or within the anterior orbit. If the cyst wall is made of epidermis without dermal tissues, it is classified as an epidermoid cyst. These lesions are occasionally lined by conjunctival or pseudostratified respiratory epithelium.17 The superficial lesions must be distinguished from deep orbital dermoids that are usually rounded, encapsulated tumors filled with fatty materials, keratin, and dermal structures such as hair particles. Histopathologically the dermoid wall is lined by keratizing squamous epithelium with dermal appendices including hair follicles and sebaceous and eccrine glands.18
Most of the dermoids are well outlined by ultrasonography because of their anterior location thus making CT or MRI rarely necessary.19 If the dermoid is unusually large or located at the frontal zygomatic suture, CT is necessary to document the relationship of the lesion to the bone before surgical intervention. Rarely, dermoids at the fronto-zygomatic suture may develop dumbbell-shaped lesions partially within the orbit and partially extending into the temporal fossa.20,21 Unusually large superior orbital dermoids particularly those that leak and create granulomatous reaction within adjacent soft tissues may erode the bone and extend into the frontal sinus or the cranium (Fig. 2).
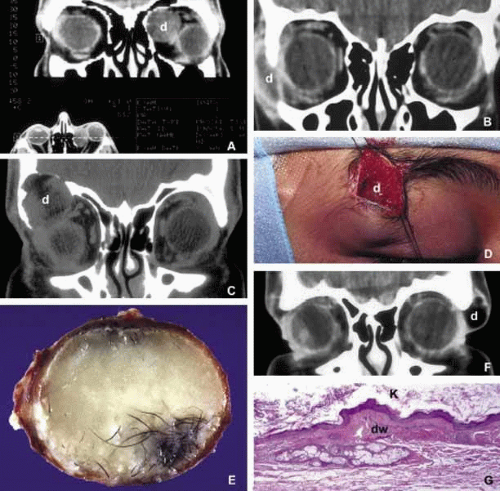 Fig. 2. Dermoid. Different presentations of dermoid (d): superior medial, semi-solid mass pushing the globe down and out (A); a ruptured dermoid causing an inflammatory reaction within adjacent soft tissues (B); a large superior lateral dermoid eroding through the roof of the orbit to extend into the brain (C); extraorbital dermoid within the subcutaneous tissues of the eyebrow (D); gross appearance of the cystic dermoid containing whitish yellow cheesy keratin material intermixed with hair (E); dumbbell dermoid that is present on both sides of the frontozygomatic fissure (F); histopathology of dermoid wall (dw) containing skin appendages, the lumen of the dermoid is lined with stratified squamous epithelium producing keratin (K) (G). ([E] is the courtesy of Amin M. Nasr, MD of Beirut, Lebanon) |
ORBITAL TERATOMA
Teratoma is a germ-cell tumor that contains tissues derived from endoderm, ectoderm, and mesoderm22 (Fig. 1). Therefore, these lesions may contain skin, bowel, lung, brain, thyroid, cartilage, and bone tissues. Most teratomas develop unilaterally and in girls. A majority of these congenital tumors are benign. Occasional reports have documented malignant transformation within orbitocranial teratomas.23 However, these benign tumors continue to grow after birth because of the collection of secretions from different tissues into the partially cystic spaces of the tumor. Some teratomas create massive proptosis and most can only be treated by exenteration. However, some of these lesions have recently been reported to be removed surgically with preservation of the globe and other vital orbital structures.
TRAUMA
Mechanical Injury
Orbital injuries result from the absorption of kinetic energy that occurs whenever the orbital tissues contact an object moving at a different speed.25 The orbital rim is capable of absorbing a considerable amount of kinetic energy without being fractured. Yet, a variety of impact forces striking the orbit may result in fractures in different areas.26 The absorption of the kinetic energy by an orbital bone may lead to contusion and/or laceration of the skin and superficial soft tissues, local deformation of the adjacent structures, globe, orbital soft tissues and bones and increases pressure in the orbital cavity. A common end result of an orbital impact is the fracture of the floor and/or the medial wall (lamina papyracea)27 (Fig. 3). Fractures of other orbital bones occur less often. Foreign bodies may be introduced into the orbit at the time of injury and may cause secondary problems depending on the nature and the location of the foreign body.28 Some foreign bodies such as copper may cause tissue necrosis and degeneration (chalcosis), and others particularly organic matter, may carry organisms such as bacteria and fungi into the orbital tissues and cause secondary infections29 (Fig. 3). Once the fracture of an orbital bone occurs, it may produce sharp edges to lacerate adjacent soft tissue structures including the globe, optic nerve, other nerves, muscles, and vessels.30 Depending on the damage of the particular tissue, functional deficit results.
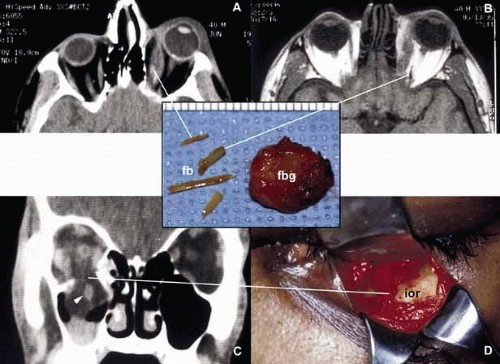 Fig. 3. Orbital trauma. Axial CT scan (A) and T1-weighted MRI (B) show multiple organic foreign bodies (fb) within the left medial rectus muscle and posterior orbit. The round tissue depicted in the inset was removed at the time of surgery; histopathologically it proved to be a foreign body granuloma (fbg). A coronal CT scan (C) and the intraoperative photograph (D) depict a large, inferior orbital rim (ior) fracture. The right inferior rectus muscle that was prolapsed into the maxillary sinus is highlighted with an arrowhead (C). (A and B are the courtesy of J. Christopher Fleming, MD of Memphis, TN) |
Another issue to deal with in an injured orbit is the development of hematoma, hematic cyst, and cholesteotoma. Hemorrhage in the orbit may occur spontaneously without any physical exertion in healthy individuals. Although terminology is not very strict, hematoma usually refers to a localized collection of blood within orbital soft tissues that develops secondary to trauma. When the blood collection within the orbit becomes organized and surrounded by a thin pseudocapsule, it is known as a hematic cyst31 (Fig. 4). If the hemorrhage develops within an existing lymphatic or vascular tumor, these lesions are known as blood cysts or “chocolate” cysts.32
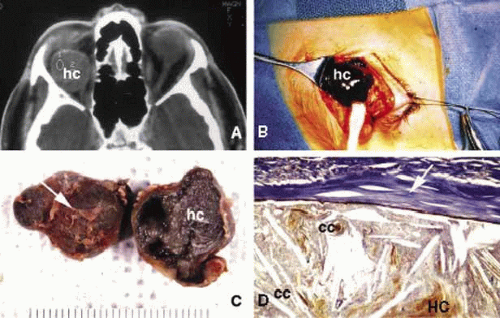 Fig. 4. Hematic cyst. Axial CT scan (A) showing a large superiorly located, well-circumscribed hematic cyst (hc) presenting as an homogeneous low-density image. Intraoperative photograph of the same case shows dark brown hematic cyst. (B). C and D show the gross and histopathologic appearance of the hematic cyst respectively. It is surrounded by a fibrous pseudocapsule (arrows) containing a mixture of cholesterol crystals, (cc), hematoidin crystals (HC), and other proteineceous debris. |
Hematic cyst consists of a localized collection of blood surrounded by a nonepithelium-lined thin fibrous capsule.33 These lesions usually develop within 1 to 2 weeks of orbital trauma but chronic cases may occur up to 20 years after orbital injury.34,35 They may reach to a size causing proptosis, extraocular motility disturbance, compression on the globe and optic nerve, that can easily be detected with ultrasonography, CT, or MRI. Hematic cysts may develop within the muscle cone or in the extraconal orbital locations.33,34,35,36 These cysts are lined by fibrovascular tissue at the periphery and contain degenerated erythrocytes, protein debris, and cholesterol crystals. In many instances the thin nonepithelial lining is adherent to the adjacent structures with fibrous tissue.
Cholesteatoma is another cystic lesion that is confined within a “pseudowall” without an epithelial lining.37 Cholesteotomas are usually located in the superior lateral orbit within the lacrimal gland fossa. Imaging studies may show a cystic, semi-cystic, or a solid lesion within the diploe of the bone or within the orbital soft tissues, with or without erosion of the adjacent bone.38 Histopathologically the lesion is composed of cholesterol clefts, hemosiderin, and hematoidin granules, other blood breakdown products and fibrin surrounded by a mixed lymphohistiocytic infiltrate and multinucleated foreign body giant cells.39
On imaging studies these lesions appear as unilocular rounded masses with destruction of the adjacent frontal and zygomatic bones. Although bone involvement in general implies malignancy, the sclerosing character of the bony destruction in choleosteoma, which is best seen in bone window images, favors a benign lesion. Although bone destruction also makes one think along the lines of metastatic tumors, one should also consider benign lesions such as brown tumor, aneurysmal bone cyst, and ruptured dermoid. Multiple cuts of the frontal bone should be examined to rule out the possibility of intracranial extension.
Osteomyelitis of the orbital bones evolving as a complication of paranasal sinusitis is another entity that should be considered in the differential diagnosis of cholesteotoma. In osteomyelitis the bone infection extends into the periosteal space and beyond. Precise delineation of the lesion can be performed with CT and MRI particularly in combination with bone SPECT, a sensitive technique used to detect osteomyelitis within cranial and orbital bones.40
Mucocele
Although a commonly encountered space occupying lesion in the orbit, mucocele is technically not a neoplasm. It is a cystic cavity lined by pseudostratified respiratory epithelium prolapsing into the orbit from a paranasal sinus, most commonly the frontal followed by the ethmoidal sinus (Fig. 5). Primary mucoceles develop as a result of an inflammatory obstruction of the ostium of the paranasal sinuses. Secondary mucoceles, however, are most commonly seen after orbital trauma and surgery; they may also develop secondary to neoplasms of paranasal sinuses and nasopharynx. If there is a superimposed infection, the lesion is referred to as pyocele. The mucocele develops as a well delineated cystic structure originating from a paranasal sinus. Depending on the location, it may compress orbital structures including extraocular muscles, optic nerve, and the globe.41 Clinical presentation of the mucocele is usually with globe displacement and/or proptosis, extraocular motility deficiency, particularly in the direction of the sinus extension into the orbit, and other compressive symptoms.42 The crepitant or calcified hard wall of the mucocele may be palpated underneath the superior or medial orbital rim. Mucoceles in general, are rare in children, however, a unique variant, ethmoidal mucopyocele, is known to occur in the medial canthal area, with lateral displacement of the globe.
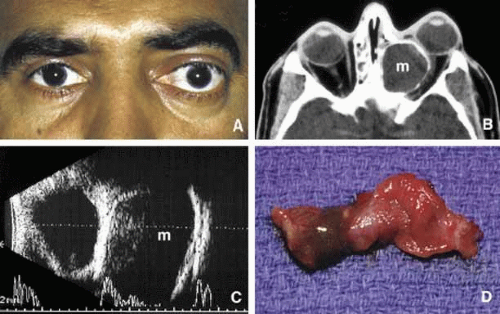 Fig. 5. Mucocele. Moderate proptosis and slight lateral displacement of the left eye secondary to a large medially located mucocele (m) originating from the ethmoid sinus (A, B). Note the compression of the calcified wall of the lesion onto the globe and the optic nerve (B). The large cystic nature of the mucocele with low internal reflectivity and segmentally calcified wall is demonstrated by ultrasonography (C). Gross specimen of the stripped mucosal lining of the mucocele from the same case (D). |
On CT, mucoceles present as hypointense, expanding masses originating from the paranasal sinuses. Early in their development these lesions are small, mucous-containing cysts. Later they are characterized by crescent-shaped and thinned remodeling of the bony walls of the orbit and sinuses.43 On MRI, mucocele presents with different appearances depending on the amount of free water within its luminal contents. When the intraluminal mucous becomes inspissated, the signal intensity in both T1 and T2 images decrease, getting closer to normal air content of the sinus.44 Treatment of mucocele is surgical excision.
VASCULAR MALFORMATIONS
Arteriovenous Fistula
Orbital arteriovenous (AV) fistulas are established as a result of abnormal flow between the arteries and veins. These lesions can be divided into three basic types: carotid cavernous, dural and orbital AV fistulas. Carotid cavernous fistula is usually traumatic but may also develop secondary to a rupture of an aneurysm particularly in elderly atherosclerotic patients. These fistulas commonly develop between an intracavernous segment of internal carotid artery and cavernous sinus and shunt arterial blood into superior ophthalmic vein.48 Dural cavernous fistulas, however, develop between small meningeal branches of internal/external carotid artery and the cavernous sinus. These small vessels that have thin walls that may rupture spontaneously particularly in hypertensive individuals, secondary to minor trauma and maintain a low blood flow.
Orbital AV fistulas usually develop secondary to traumatic rupture of the ethmoidal artery into the orbital venous system. This type of fistula maintains a low blood flow. Clinical findings of AV fistulas include rapidly developing proptosis, edema of the conjunctiva and eyelids, dilatation and tortuosity of the conjunctival and episcleral vessels, and secondary glaucoma.
Most of these cases are diagnosed with imaging procedures including CT, MRI, angiography, color Doppler ultrasonography, and catheterized angiography.49 Current treatment of these lesions is embolization via catherization.50 Morphologic data are limited to autopsy material because most patients with AV fistulas do not undergo biopsy procedure. These lesions show irregular, malformed arteries and veins with abnormal elastic and muscular layers and secondary endothelial cell proliferation. Approximately half of the low shunt fistulas close spontaneously;51 therefore, it is best to follow-up some of these patients conservatively if they do not have severe symptoms.
Orbital Varix
Orbital varix is a rare vascular lesion with questionable histopathogenesis. The absence of valves in the orbital venous system and the weakening of venous wall may lead to pooling and stasis of blood resulting in distention of the venous channel with thrombosis. In gross appearance, the varix is a distended vein containing a canalized or uncanalized thrombus.52,53 Histopathologically varix consist of irregular vascular channels lined by endothelial cells. In chronic lesions, the blood vessel walls irregularly thicken with fibrosis and deposits of chronic inflammatory cells mixed with deposits of calcium and hemosiderin pigment are seen. Orbital varices are divided into primary and secondary types. The primary orbital varix is confined to the orbit as an isolated lesion without any connection to other A-V malformations. The secondary orbital varix, however, develops as an extension of an intracranial AV malformation that shunts blood to the orbital venous system causing the venous channels to distend secondarily.54 Management of orbital varix consists of total surgical excision when possible and/or endovascular embolization.
ORBITAL INFLAMMATION
MICROBIAL INFLAMMATION
Microbial infections of the orbit develop because of a variety of organisms, which are introduced to the orbit by different routes. Bacterial orbital infections are the most common that primarily originate from the paranasal sinuses and to a lesser degree, secondary to penetrating injuries with or without foreign bodies and rarely, secondary to endophthalmitis or systemic infectious process such as bacterial septicemia, tuberculosis, etc. The most common cause of bacterial orbital cellulitis is Staphylocci and Streptococci species. Although Haemophilus influenza was a common causative organism in children younger than 4 years of age, recent vaccination efforts have decreased the incidence of H. influenza cellulitis significantly.55
Anatomically speaking, if the infection is limited to anterior of the orbital septum, involving the eyelids it manifests itself primarily with lid and conjunctival edema and some venous congestion but not with proptosis.56 This process that is known as preseptal cellulitis is usually initiated with skin injuries or an upper respiratory tract infections in children. However, if the infectious process involves orbital soft tissues behind the septum, it is called orbital cellulitis (Fig. 6). Cellulitis usually develops secondary to a paranasal sinus disease, clinically associated with axial proptosis, marked chemosis, congestion of retinal veins, limitation of extraocular motility, and swelling and pain around the eye. Depending on the severity of the condition, vital structures within the orbit such as the globe and the optic nerve may be compressed or infiltrated by the infection. Generally, in children the orbital cellulitis originates from the ethmoid sinus and may limit itself to the subperiosteal site for a while and later spread into orbital soft tissues.
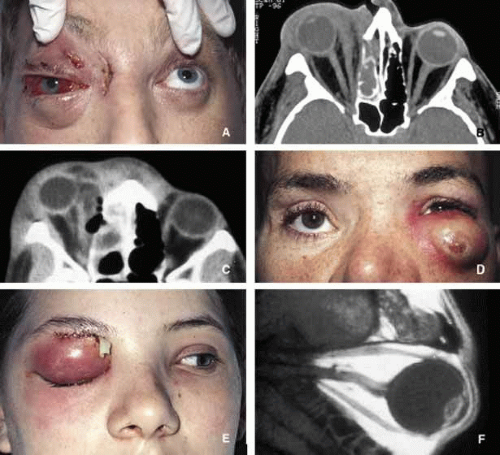 Fig. 6. Orbital cellulitis. A and B show the appearance of right orbital cellulitis secondary to Staphylococcus infection from right ethmoidal sinus. The proptosis, chemosis and visual loss in this patient worsened after the ethmoidectomy and the medial orbit had to be explored with additional drainage of pus. Note the stretch of the right optic nerve and pear-shaped right globe secondary to marked proptosis on an axial CT scan (B). C shows another case with severe ethmoiditis causing cellulitis of the right orbit with the extension of the infection preseptally toward the left globe. The patient responded well to ethmoidectomy and drainage of the abscess from the right medial orbit. D shows the appearance of a preseptal and anterior orbital cellulitis secondary to a long-standing foreign body caused in a motor vehicle accident. The patient depicted in frame E developed Streptococcal orbital cellulitis with secondary brain abscess as depicted in the sagittal T1-weighted MRI (F). |
Acute bacterial orbital cellulitis presents a nonspecific histopathology associated with diffuse polymorphonuclear infiltrate that may on occasion lead to abscess formation57 (Table 2). Subperiosteal and soft tissue abscesses may present with decreased vision, disc edema and increased intraorbital and intraocular pressures. Orbital abscess is best demonstrated with MRI. Emergent orbital exploration and the drainage of the abscess is indicated.
TABLE 2. Correlation of Cellular Pathology to Clinic Diagnosis in Orbital Inflammatory Lesions | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
IOI, idiopathic orbital inflammation (orbital pseudotumor); PMN, polymorphonuclear cell; w, with; w/o, without.
Orbital cellulitis may also be caused by different types of foreign bodies including organic and nonorganic matter and nonautogenous surgical implants.28 Inorganic foreign materials, such as metal and glass, are usually well-tolerated and do not cause infection unless they significantly distort the orbital anatomy with exposure to periorbital sinuses and nasal cavity. Organic foreign bodies such as wood, vegetable fibers, etc., however, trigger significant foreign body reaction and sometimes suppurative inflammation.These should be documented by CT and/or MRI and surgically removed29 (Fig. 3). Today many nonautogenous materials are used in ocular and orbital reconstruction, including porous implants, mesh materials, polymeric silicone plates, sponges for scleral buckling procedures and reservoirs of drainage valves for glaucoma. Implants and repair blocks made of porous materials may lead to acute infection when they erode through sinus or conjunctival epithelium. Whenever there is a foreign body, noncaseating granulomatous reaction with multinucleated giant cells is identified, adjacent to the foreign material; secondary acute inflammation may be superimposed. In penetrating injuries the nature of the foreign body is an important factor.
An exception to suppurative soft tissue reaction that is caused by bacteria is chronic caseating granulomatous inflammation, which is caused by mycobacteria and certain types of fungi (Table 2). Ocular and adnexal tuberculosis is usually seen with manifestation of systemic mycobacterial infection,58,59 Because of the recent increase in the numbers of immunologically suppressed individuals secondary to viral epidemics and wider use of immunosuppressant antimetabolites in longer-surviving cancer and transplantation patients, the incidence of tuberculosis has been increasing steadily during the past two decades and the clinical picture of the disease has been changing, with many cases developing because of atypical mycobacteria that are resistant to traditional multidrug treatment.61 It has been reported that the individuals with HIV/AIDS have an incidence of tuberculosis 500-fold more than that is seen in the general population.62
The orbital disease, is more often seen in children and nonwhite patients. History of antecedent penetrating injury is a common presentation of tuberculosis, caused by atypical mycobacteria. Histopathology of tuberculosis consists of zonal granulomatous inflammation with numerous epitheliod histiocytes surrounding a necrotic (caseating) center. Tissue diagnosis is pathognomonic only with the documentation of positive acid-fast organisms; however, in many cases special stains may fail to demonstrate the mycobacteria, but the cultures grow M. tuberculosis or atypical mycobacteria. Orbital tuberculosis is usually associated with systemic disease (Fig. 7).
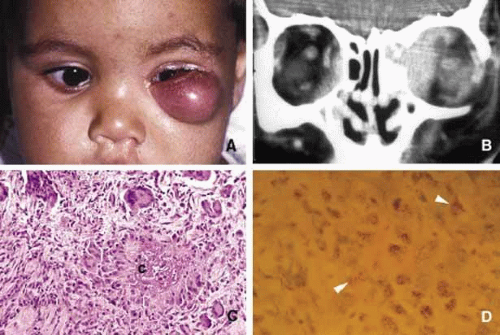 Fig. 7. Orbital tuberculosis. A 2-year-old boy with left orbital tuberculosis (A). Both mother and child had systemic disease. A case of bilateral orbital and perinasal sinus tuberculosis is shown in the coronal CT of a 28-year-old man (B). Note the irregular involvement of the bony tissues of the orbits and the sinuses. C and D show a caseating granuloma (c) and AFB-positive tuberculous bacilli in necrotic inflammation (arrowheads) respectively. |
In most instances, fungi infect the orbit as an extension of paranasal sinus disease or after penetrating injury associated with the introduction of organic matter. Most of the fungal infections, particularly mucormycosis often develop in immunocompromised patients.63
Orbital mucormycosis is an emergency situation because it causes rapidly progressing necrotizing inflammation secondary to vascular involvement (Fig. 8). Orbital exploration should be performed immediately to establish the diagnosis by identifying the broad, nonseptated hyphae and for surgical debridement as well as irrigation with antifungal agents. The prognosis of mucormycosis is very poor. Aspergillosis, unlike mucormycosis, presents a low-grade, smoldering chronic granulomatous inflammation that may be confused with primary orbital tumor.64 The identification of the organism in fungal and parasitic disease, is crucial at the time of surgery; fungi may also be identified with smears and frozen sections.65 In any kind of orbital exploration secondary to cellulitis, tissue samples should be obtained for Gram, fungal and AFB stains and aerobic, anaerobic and fungal cultures.
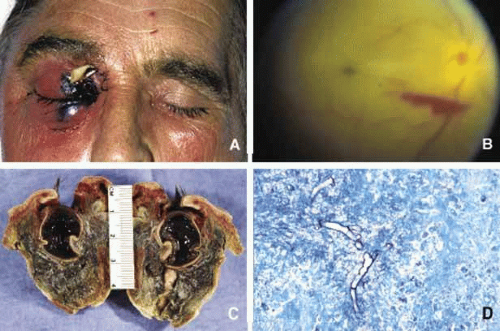 Fig. 8. Mucormycosis. The appareance of mucormycosis in the right orbit, periorbital skin and maxillary sinuses of a 60-year-old man with diabetic ketoacidosis (A). B shows the funduscopic apperance of a central retinal artery thromboembolism with resultant “cherry-red spot” from another case of orbital mucormycosis. C shows the extensively necrotic, bloodless cut surface of the exenteration specimen from the patient shown in A. Histopathologic examination of this specimen revealed numerous nonseptated mucormycosis hyphae with 90-degree branching (D) in Gomori methanamine silver (GMS) stain. |
As a rule, no matter how rarely, any microbial inflammation, including bacteria, fungi, viruses, and others, may infect the orbit and cause acute, chronic or granulomatous inflammation leading to tissue damage and fibrosis. These rare diseases include syphilis,66 leprosy,67,68 Parinaud syndrome,71,72 and actinomycosis.73,74 Examples of parasitic orbital infections include echinococcosis, cysticercosis, myiasis, and trichinosis of extraocular muscles.76,77,78,79,80,81
NONMICROBIAL INFLAMMATION
Many forms of inflammatory processes may trigger orbital inflammation that simulates neoplasms by producing proptosis and associated orbital findings.82 These include Graves disease,83,84,85 idiopathic orbital inflammation (orbital pseudotumor),86,87 Tolosa-Hunt syndrome,88,89 sarcoidosis,90,91 Sjögren syndrome,92,93 and Wegener granulomatosis.94,95,96
Graves Disease
Thyroid-associated orbitopathy, better known as Graves disease (Gd), is an idiopathic orbital inflammation that primarily involves the muscles and soft tissues of the orbit and the eyelids. The commonly involved muscles include inferior, medial, superior and lateral recti that cause swelling of the tissue leading to proptosis and eyelid retraction (Fig. 9). Gd is the most common cause of unilateral and bilateral proptosis in adults; although uncommonly it may be seen in children as well.
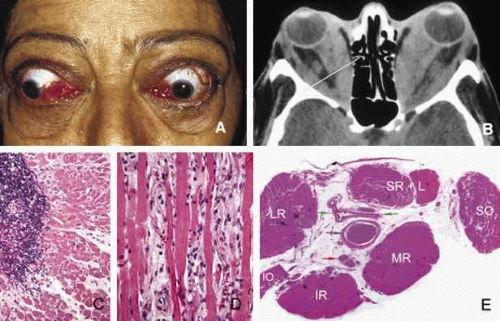 Fig. 9. Graves disease. Clinical findings of Graves disease are depicted in frame A, including bilateral proptosis and lid lag with extraocular motility disturbance, chemosis with prolapse of lacrimal gland and congestion of conjunctival and episcleral blood vessels. Axial CT scan (B) reveals marked swelling of all recti muscles. The histopathologic appearance of the extraocular muscle from a patient with Graves disease reveals chronic inflammatory infiltrates, primarily composed of lymphocytes and plasma cells (C). The extraocular muscle volume is increased because of diffuse endomysial fibrosis, mucopolysacccharide deposition and chronic inflammatory cell infiltration (D). The orbital fat, meninges and the optic nerve (blue arrow), large blood vessels of the orbit, such as ophthalmic artery and its branches (green arrows) and the ciliary ganglion (red arrow) do not show any inflammation. The enlargement of the extraocular muscles are well depicted in frame E, which represents a transverse section, approximately at the level indicated by the yellow line in frame B. (LR: lateral rectus; SR + L: superior rectus and levator complex; SO: superior oblique; MR: medial rectus; IR: inferior rectus; IO: inferior oblique). (Frames C, D, and E are the courtesy of Ralph C. Eagle, MD of Philadelphia, PA) |
The pathogenesis of Gd is not completely understood; therefore, it is labeled as an “autoimmune” process.82,98 It has been suggested that individuals with HLA-B8 major histocompatility antigen Haplotype are genetically susceptible to Gd.99 The hypothesis is that circulating T-cells directed against an antigen in thyroid follicular cells recognize a similar antigen in extraocular muscles and orbital soft tissues.100 Experimental studies suggest that thyrotropin receptor (TSH-R) is one of the possible entities to stimulate the autoantibodies that lead to the inflammatory changes within orbital soft tissues. In Gd there is a predominance of T-cells with Th1 profile, although Th2 profile of cytokine production has also been reported.100 The cytokines stimulate fibroblasts to produce glycosaminoglycans that in turn lead to deposition of this substance within the muscle tissue leading to anatomic and functional deficiencies (Fig. 9). Although cell mediated immune reaction predominate in early Gd, humoral immunity plays a greater role in later phases.101
Some of the immune changes are reflected in the histopathology of Gd, which can basically be divided in two stages. The active inflammatory stage consists of perivascular edema and clustering of lymphocytes and plasma cells; lymphoid follicles are not frequent in Gd. In the chronic stage the volume of the involved orbital tissues are increased because of the deposition of glycoproteins and mucopolysaccharides and secondary to the infiltration of fibroblasts producing collagen. Later in chronic stages of the disease, the edema decreases and the muscles are primarily infiltrated with interstitial fibroblasts and chronic inflammatory cells leading to fibrosis.
Although 80% of patients with Gd present with a history of hyperthyroidism, approximately 10% suffer hypothyroidism or autoimmune thyroiditis; occasionally, an euthyhroid individual may also develop the signs and symptoms of Gd.102 Upper lid retraction is the most frequent clinical sign in early Gd (75%) followed by asymmetrical bilateral proptosis (60%) and restriction of extraocular muscles (40%). Compressive optic neuropathy may result secondary to the enlargement of the extraocular muscles in the apex. Optic nerve malfunction is manifested by afferent pupillary defects and color vision and visual field deficiencies in approximately 5% of Gd patients.
The best means of evaluation of extraocular muscles is imaging with CT and/or MRI.11 Axial CT scan is very valuable to depict the enlargement of the extraocular muscles and determine whether there is any infiltration into other orbital soft tissues. Coronal sections are also very useful to evaluate the enlargement of the muscles and their relationship to the optic nerve in the orbital apex.103 In differential diagnosis of Gd, one should keep in mind that it is not only the most frequent cause of bilateral proptosis but unilateral proptosis as well. Therefore, the slowly progressive unilateral presentation may be confused with orbital pseudotumors, neoplasia, and solitary vascular lesions such as orbital varix.104,105 Orbital metastatic neoplasms may also be confusing if they are limited to the extraocular muscles. Imaging usually reveals nodular enlargement of the muscle and the diagnosis of a metastatic disease may be confirmed with fine needle aspiration biopsy (FNAB). The treatment of Gd includes oral and intravenous steroids, radiation and surgery.
Idiopathic Orbital Inflammation (IOI)
Orbital pseudotumor is a nonspecific chronic inflammatory condition of unknown etiology. Although an underlying immune process is suspected, no conclusive mechanism has been established for the development of this curious entity.87 IOI may develop with sudden onset of painful proptosis associated with motility disturbances, eyelid swelling, redness and chemosis. It may develop as a diffuse or localized lesion and its histopathology varies accordingly from case to case; the histopathology is also variable at different stages of the disease. The extraocular muscles may be involved with the inflammatory process but the main target is the orbital fibroadipose tissue. The inflammatory process may be grouped into two main categories: (1) diffuse and (2) localized nonspecific orbital inflammation. The localized nonspecific inflammation is further divided according to specific sites, i.e., myositis, dacryoadenitis, periscleritis and perineuritis. Each of these subgroups may present as an acute, subacute or chronic process in a given patient.
The histopathology of the IOI usually consists of a mixed polymorphonuclear and lymphocytic infiltrate during the early phases; as the disease advances, lymphoid follicle formation and fibrous tissue proliferation dominates the picture106 (Fig. 10). Patchy aggregates of lymphocytes and/or lymphoid follicles are frequently seen, but these lymphoid aggregates are not confluent as in lymphoid neoplasia and hyperplasia. Because of the diffuse fibrous reaction and the nonspecific nature of the mixed inflammatory infiltrate, the biopsy diagnosis of pseudotumor is not pathognomonic, but it should be correlated with clinical and radiologic findings in each case. Histopathologic patterns may vary in different regions of the specimen, therefore, it is advisable that these biopsies are processed totally. Hard granulomas, perivascular lymphocytic infiltrates, and occasionally true vasculitis may be identified within the orbital tissues of clinically typical IOI. Although polymorphonuclear leukocytes and eosinophils are occasionally seen, prominent acute inflammatory infiltrates should lead the pathologist to consider a vasculitis, such as polyarthritis nodosa or Wegener granulomatosis.
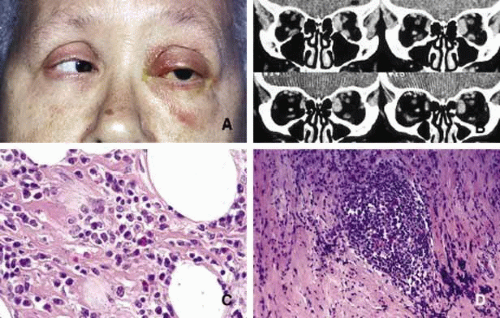 Fig. 10. Orbital pseudotumor. A patient with subacute orbital pseudotumor showing mild proptosis with hyperemia and edema of the eyelids and limited extraocular motility of the left eye (A, B). The left medial rectus muscle is severely involved with the disease that is also present to a lesser degree within the left inferior rectus muscle. Frames C and D reveal a mixture of lymphocytes, plasma cells, and eosinophils wthin fibroadipose tissues of the orbit. |
Tolusa-Hunt syndrome, otherwise known as painful external ophthalmoplegia, is another inflammatory process of the orbit of unknown etiology. It is conceivable to think that it represents a localized form of idiopathic orbital inflammation (IOI) and presents with typical clinical manifestations because of its presentation in the orbital apex. The clinical symptoms include a severe, constant deep orbital pain associated with functional deficiencies of third, fourth, fifth and sixth cranial nerves.107 Typically, the orbital pain that presents abruptly also responds to systemic corticosteroid treatment with the same abruptness. Other symptoms of the disease including third, fourth, and sixth cranial nerves palsies and the hypesthesia of the periorbital skin also respond well to corticosteroid treatment. Although bilateral cases do occur; a great majority of patients with Tolosa-Hunt syndrome present unilaterally and therefore, should be differentiated from the tumors, which may involve the orbital apex including meningioma, pituitary adenoma, neurofibroma, paraganglioma, nasopharyngeal squamous cell carcinoma and metastatic tumors.108,109
Tumors of the apex, however, usually cause a gradual development of motility dysfunction depending on the location of the tumor that may be accompanied with dull pain but usually not with an abrupt onset of panophthalmoloplegia and explosive pain.
Nonspecific Granulomatous Inflammation
Sarcoidosis is a multisystem disease of an idiopathic nature that commonly involves the orbit and the eye. Systemically it involves the lungs and the upper respiratory tract, liver, spleen, lymphatic and hematopoetic tissues, central nervous system and the skin. Although there is considerable evidence in favor of sarcoidosis being infectious in nature, no causative agent has been established so far. The etiopathogenesis of sarcoidosis is still unknown.110 The typical noncaseating granulomas are made of T-lymphocytes of helper and suppressor types and dendritic Langerhans cells with human leukocyte antigen (HLA)-DR expression. Perivascular inflammatory reaction is characteristic of a delayed type hypersensitivity response. Some investigators think that noncaseating epithelioid granulomatous reaction, which is determined as the hallmark feature of the disease may be the host’s immune response to presently unknown causative agent(s). Although the exact significance of granuloma formation in sarcoidosis is not clearly known, it appears that this tissue reaction is a secondary event as a result of exaggerated cellular immune response to a class of unknown antigens. It is hypothesized that the suppressor aspect of cell mediated response in sarcoidosis is abnormal and therefore reduces the function of helper T-lymphocytes. The initial step in granuloma formation of sarcoidosis is considered to be triggered by a cytokine (interleukin-1) that increases the proliferation of helper T-lymphocytes and activate these cells. Activated helper T-cells in turn secrete interleukin-2; a mitogen, which stimulates the proliferation of helper T-cells even further.111 As a consequence these cells aggregrate at the site of the causative insult and secrete monocyte chemotactic factors that lead to the gathering of epitheloid macrophages and multinucleated giant cells to form granulomas. Sarcoidosis is also associated with increased B-cell activity manifested by polyclonal hyperglobulinemia.112,113,114
Sarcoid granulomas are made of epithelioid cells and multinucleated giant cells, surrounded by lymphocytes and occasional plasma cells (Fig. 11). Many inclusion bodies have been described in the giant cells of sarcoidosis but none of these is pathognomonic. The granulomatous response of sarcoidosis is rather typical but not unique for this entity; fungal diseases, tuberculosis, Crohn disease and leprosy may produce similar granulomas115 (Table 2).
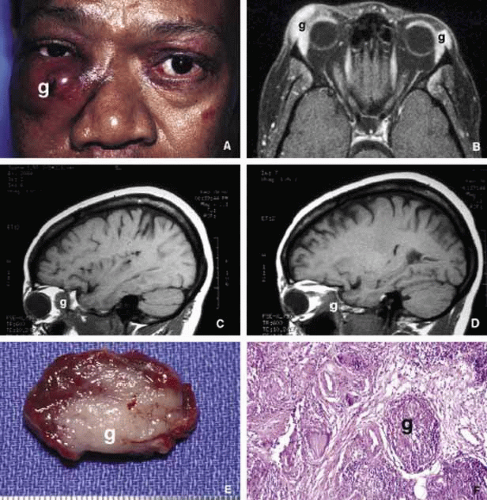 Fig. 11. Sarcoidosis. The appearance of a large sarcoidosis granuloma involving the anterior orbit and periorbital skin (g). The central dome-like lesion is caused by secondary to a Staphylococcus infection. In addition, plaquoid skin lesions of sarcoidosis are seen above the left eyebrow, on the left upper eyelid, and on the periorbital skin (A). The T2-weighted axial MRI with contrast reveals bilateral enlargement of lacrimal glands (g) in another case B. Frames C and D show T1-weighted sagittal MRIs with localized granulomatous masses of sarcoidosis within the posterior orbit involving the apex and extending into the cavernous sinus (C, D). A well-delineated but not encapsulated mass of a sarcoid granuloma (E). The histopathology of sarcoidosis consists of multiple granulomas composed of histiocytic cells, chronic inflammatory cells, and multinucleated giant cells (g) (F). |
Approximately one-fourth of sarcoidosis patients develop ocular and orbital manifestations including anterior and posterior uveitis, chorioretinitis, conjunctival and eyelid granulomas and orbital mass lesions (Fig. 11). Lacrimal gland is a common site of involvement; autopsy studies show a high percentage of microscopic disease; however, only 15% to 20% of the patients show clinical symptoms. Although virtually any part of the orbit may be involved with sarcoidosis, the most common site is the lacrimal fossa and the disease in this location may be confused with chronic dacryoadenitis, Sjögren syndrome, or space-occupying lesion. Sarcoid granulomas may also extend into the orbit from adjacent sinus mucosa.116 If other manifestations of the disease are absent, these cases may mimic secondary orbital tumors and they can only be differentiated by biopsy.
Patients with distinctive systemic manifestations with bilateral hilar lympadenopathy, skin lesions, uveitis, etc., usually show increased angiotensin-converting enzyme (ACE) levels.117 Serum lysozyme and calcium levels may also be increased in sarcoidosis but neither one of these tests is specific for the disease. The ultimate diagnosis is by biopsy. Some advocate to perform biopsy only on the sarcoid-suspect lesions such as skin and conjunctival nodules in which the yield is usually rewarding. Others support random “blind” biopsy of the conjunctiva in sarcoid suspects. The yield of random biopsy without a distinct lesion is rather low (approximately 25% positive), but the conjunctival biopsy carries low morbidity and can be inexpensively and quickly performed in the clinic, as opposed to more invasive biopsies of transbroncheal lymph nodes, liver, and orbit. It is advisable, from the practical standpoint, to biopsy the conjunctiva randomly early in the workup of a sarcoidosis-suspect patient.118 If the biopsy result reveals granulomatous inflammation, more invasive procedures with high morbidity and cost can be avoided.
The involvement of the optic nerve with sarcoidosis is usually an anterior process and associated with typical retinal vasculitis, however, the optic nerve involvement may rarely extend posteriorly and form a mass lesion.119
The treatment of sarcoidosis is directed to the systemic disease. Surgery may be necessary to biopsy or debulk orbital lesions if there is a need for histopathologic evaluation in patients with no other easily accessible biopsy sites. In few instances, the orbital disease presents with no history or detectable symptoms of systemic sarcoidosis (Fig. 11). In these patients, sarcoidosis is usually a surprising diagnosis obtained from an orbital “tumor.” The mainstay of treatment is the use of systemic corticosteroids and antimetabolites such as methotrexate.
Sjögren syndrome (SS) consists of a triad of symptoms including dry eyes (keratoconjunctivitis sicca), dry mouth (xerostomia), and “dry joints” (arthritis).120 Primary SS is not associated with other connective tissue diseases; however, secondary SS symptoms overlap with the manifestations of systemic lupus erythematosis, polymyositis, polyarteritis nodosa, scleroderma, and rheumatoid arthritis.121 Like many other autoimmune diseases, SS does not have a clear cut etiology, however, primary SS is considered a mononuclear inflammatory vasculopathy closely linked to HLA-DR3 and HLA-DRw52; secondary SS associated with rheumatoid arthritis is linked to HLA-DR4.122 Many viruses, including Epstein-Barr, CMV, HIV, and hepatitis-C, have been reported to have an etiologic role in SS. Immune complex formation and deposition are considered to be the physiopathology of cutaneous and ocular vasculitis.123
Histopathology of the conjunctiva as well as the lacrimal gland is nonspecific consisting of lymphocytic and plasma cell infiltrates surrounded by eosinophilic basement membrane like material (Table 2). These units are called epimyoepithelial islands and are considered to be diagnostic of SS.124 Lacrimal gland also reveals acinar atrophy and increased fibrosis surrounding the ductules125 (Fig. 12). Diagnosis of SS is based on minor salivary gland biopsy rather than the biopsy of the lacrimal gland, because the latter procedure is more invasive and carries a higher morbidity.126
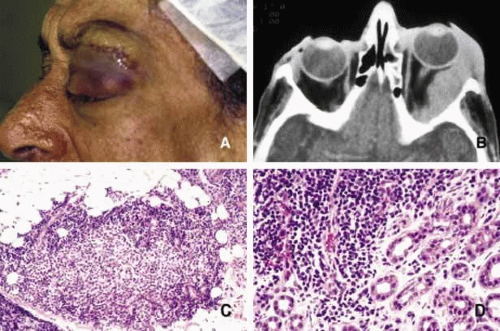 Fig. 12. Sjögren disease. A pateint with bilateral enlargement of lacrimal glands that was involving the left side more than the right (A,B). Minor salivary gland biopsy from the lower lip revealed infiltration of eosynophils, lymphocytes and plasma cells. The biopsy was sufficient to make the diagnosis combined with the clinical picture. The same patient developed orbital lymphoma, which is depicted in frame B, 3 years after the diagnosis of Sjögren disease. |
Keratoconjunctivitis sicca is the most common presentation of SS in the eye occurring in approximately 90% of patients. Diminished tear meniscus and decreased tear break up time (BUT) with diminished tear production documented with Schirmer strips are common findings. Less commonly, patients develop episcleritis/scleritis in primary type of SS.127 Because of peripheral and central nervous system involvement, optic neuritis and internuclear ophthalmoplegia may be seen in these patients. From the orbital standpoint, the asymmetrical presentation of the disease may be confused with an orbital lymphoma or sarcoidosis. In most cases, however, the disease presents with bilateral enlargement of the lacrimal glands and with the presence of other symptomatology SS is easy to diagnose (Fig. 12). It should be kept in mind, however, that SS patients have an increased risk of developing B-cell lymphomas in the salivary glands and cervical lymph nodes. This association was not found to be true for the lacrimal gland. However, orbital lymphoma that may mimic the presentation of SS should always be considered in the differential diagnosis.
Wegener granulomatosis (WG) is an idiopathic systemic vasculitis that also causes necrotizing granulomatous inflammation.128 The classical triad of the disease includes necrotizing granulomatous vasculitis of upper and lower respiratory tracts, and necrotizing glomerulonephritis. Small vessel disease also affects the eye and orbit leading to conjunctivitis, scleritis, uveitis and thromboembolic phenomenon of the choroidal vessels and central retinal artery.94,95,129
Orbital involvement also results from necrotizing vasculitis with or without granulomatous inflammation leading to painful proptosis, eyelid and conjunctival edema, and extraocular motility disturbance. Optic nerve disease may result from the combination of vasculitis of the optic nerve and meningeal vessels and/or the compression caused by an orbital space occupying lesion.95 This on occasion may lead to occlusion of the central retinal artery.130
Although the specific pathogenesis of WG is unknown, there is consensus that the disease develops as a result of an autoimmune mechanism, however, it does not appear to be caused by immune complex deposition like other forms of vasculitis.131 It has been speculated that the vasculitis of WG is triggered by an infectious process.132,133 As a rule, the respiratory tract involvement in WG precedes renal or systemic disease, however, many atypical cases with lack of involvement of one organ system or another are well recognized.134
It is well known that anti-neutrophil cytoplasmic antibodies (ANCA) function to contain the inflammatory responses by proteolysis, primarily by collagenases and elastases.135 C-ANCA is a very sensitive and specific serologic marker for WG with a sensitivity increasing up to 96% for active disease.136,137 Others hypothesize that C-ANCA is not merely a marker for the disease but in itself is pathogenic.138 High and low C-ANCA titers are known to correlate well with disease activity and remission respectively.139,140 Biopsy-proven head and neck particularly orbit cases are known to occur without elevated titers of C-ANCA.141
The classical histopathologic picture includes necrotizing vasculitis with necrosis and granulomatous inflammation (Table 2). However, a considerable variability is observed in the biopsies obtained from different organs and the classical appearance is not always demonstrable.142,143 Upper respiratory tract and orbit biopsies usually show vasculitis and necrosis but rare granulomas.144,145 Lung biopsy results usually present with diffuse necrotizing vasculitis of small blood vessels resembling an infectious process. Polymorphonuclear cells and eosinophils may form cuffs around blood vessels but granulomas are rare.146 Kidney biopsies show necrotizing glomerulonephritis without well-formed granulomas.147
Orbital biopsy samples also fail to depict the typical combination of vasculitis and granulomatous inflammation (Fig. 13). Kalina and co-workers reported the presence of complete triad of vasculitis, necrosis and granulomatous inflammation in only 54% of the biopsies.142 Granulomas also present some variability; in certain instances they are seen as typical hard granulomas made of aggregates of epitheloid cells, and giant cells surrounded by lymphocytes and occasional plasma cells. Granulomas that present within necrotic areas, however, may present as palisading lesions containing numerous polymorphonuclear leukocytes and eosinophils.
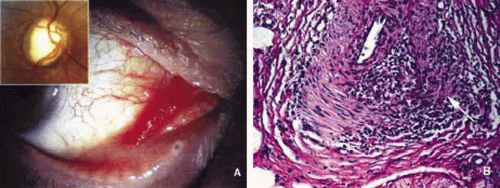 Fig. 13. Wegener granulomatosis. Mild proptosis combined with conjunctival chemosis and hyperemia adjacent to congested tortuous blood vessels of a Wegener granulomatosis patient. The right optic nerve disc shows optic nerve pallor (inset) (A). The excisional biopsy from the conjunctival lesion reveals granulomatous vasculitis with focal necrosis (arrow) (B). |
Ophthalmic involvement of WG is best categorized in two types: (i) focal disease that is the result of vasculitis and primarily affects the anterior and posterior segments of the eye and (ii) contiguous disease that is primarily seen in the orbit as a result of WG extending from the nasal cavity and perinasal sinuses. Orbital disease that develops acutely with painful proptosis, eyelid and conjunctival edema and ocular motility disturbance is the most common ocular manifestation in WG 95,148 (Fig. 13). This presentation of WG may mimic orbital pseudotumor and infectious cellulitis as well as lymphoma and metastatic carcinoma.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


