Purpose
To compare the efficacy and safety of the European labels of ranibizumab 0.5 mg vs dexamethasone 0.7 mg in patients with macular edema secondary to central retinal vein occlusion (CRVO).
Design
Phase IIIb, multicenter, double-masked, randomized clinical trial.
Methods
Patients were randomized (1:1) to receive either monthly ranibizumab followed by pro re nata (PRN) treatment (n = 124) or 1 sustained-release dexamethasone implant followed by PRN sham injections (n = 119). Main outcomes were mean average change in best-corrected visual acuity (BCVA) from baseline to month 1 through month 6, mean change in BCVA, and adverse events (AEs).
Results
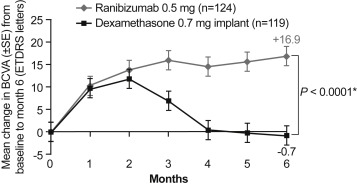
Of 243 patients, 185 (76.1%) completed the study. No difference was observed in BCVA between ranibizumab and dexamethasone at months 1 and 2. From month 3 to month 6, there was significant difference in BCVA gains in favor of ranibizumab. At month 6, mean average BCVA gain was significantly higher with ranibizumab than with dexamethasone (12.86 vs 2.96 letters; difference 9.91 letters, 95% confidence interval [6.51–13.30]; P < .0001). Mean injection number of ranibizumab was 4.52. Ocular AEs were reported in more patients in the dexamethasone than in the ranibizumab group (86.6% vs 55.6%).
Conclusions
Using the European labels, similar efficacy was observed for ranibizumab and dexamethasone at months 1 and 2. However, ranibizumab maintained its efficacy throughout the study, whereas dexamethasone declined from month 3 onward. The main limitation of the study was that dexamethasone patients received only a single treatment during the 6-month study. In clinical practice, dexamethasone retreatment may be required earlier than 6 months. Safety findings were similar to those previously reported.
Macular edema secondary to central retinal vein occlusion (CRVO) is a common retinal vascular disease associated with rapid vision loss. Two treatment options—anti–vascular endothelial growth factors (anti-VEGFs) and corticosteroids—with different mechanisms of action are available for the treatment of macular edema secondary to CRVO. Ranibizumab (Lucentis; Novartis Pharma AG, Basel, Switzerland, and Genentech Inc, South San Francisco, California, USA), a humanized monoclonal antibody fragment (Fab), was the first anti-VEGF agent approved for the treatment of macular edema secondary to CRVO.
Ranibizumab 0.5 mg was approved in June 2010 by the United States Food and Drug Administration (US FDA) based on the 6-month results of a phase III, randomized, double-masked controlled trial—Central Retinal vein occlUsIon Study: Evaluation of efficacy and safety trial (CRUISE). In the CRUISE study, a statistically significant improvement in the mean best-corrected visual acuity (BCVA) was observed with monthly ranibizumab 0.5 mg injections vs sham at month 6 (14.9 vs 0.8 letters, P < .0001), with an average of 5.5 intravitreal injections. In addition, at month 6, a significantly greater proportion of patients from the ranibizumab group gained ≥15 letters compared with those from the sham group (47.7% vs 16.9%, P < .0001). During the 6-month pro re nata (PRN) treatment period of the CRUISE study, improvements observed with monthly dosing during the first 6 months were sustained over 12 months after switching to an individualized, as-needed dosing regimen guided by BCVA and central subfield thickness retreatment criteria. HORIZON and RETAIN, the subsequent extension studies, established long-term safety and efficacy of ranibizumab 0.5 mg over a period of 4 years, with no new safety concerns and maintenance of visual acuity (VA) gain in CRVO.
Dexamethasone intravitreal implant (Ozurdex; Allergan, Inc, Irvine, California, USA), a potent water-soluble biodegradable corticosteroid, is an anti-inflammatory agent that can be delivered to the vitreous cavity. Dexamethasone intravitreal implant was approved in June 2009 by the US FDA for the treatment of macular edema secondary to CRVO based on the 6-month results of a phase III sham-controlled study—Global Evaluation of implaNtable dExamethasone in retinal Vein occlusion with macular edema (GENEVA). In the GENEVA study, a significant improvement in BCVA with dexamethasone intravitreal implant vs sham was achieved from day 30 to day 90 (greatest difference at day 60: approximately 9 letters; P < .001) following 1 intravitreal application for macular edema due to BRVO and CRVO. In addition, a greater proportion of patients with CRVO showed an improvement of ≥15 letters with dexamethasone intravitreal implant compared with sham at day 180 (18% vs 12%), with the greatest response at day 60 (29% vs 9%, P < .001). The GENEVA study was unable to determine the duration of effect of the dexamethasone intravitreal implant or the optimal retreatment interval. However, 12-month data showed that repeated dexamethasone intravitreal implant application was associated with repeated but temporary visual improvement and steroid-related complications such as elevated intraocular pressure (IOP) and cataracts.
The optimal retreatment interval for the dexamethasone intravitreal implant remains unclear. The retreatment protocol in the GENEVA study did not address the possibility of patients requiring treatment earlier than 6 months: there were no planned visits in the study between months 3 and 6. Furthermore, some smaller studies, including a recent post hoc analysis, have shown that the efficacy of dexamethasone intravitreal implant does not last over 6 months. The post hoc analysis by Kuppermann and associates showed that the effect of the dexamethasone intravitreal implant in terms of BCVA improvement started at day 7 and lasted for 2–3 months when considering >3-line gainers. Recently published studies have investigated the use of the dexamethasone intravitreal implant at shorter intervals (4–6 months) than those proposed in the dexamethasone intravitreal implant label, with better anatomic and functional outcomes.
Separate clinical trials have shown both ranibizumab and dexamethasone intravitreal implant to be effective in improving VA in patients with CRVO, but head-to-head trials directly comparing the efficacy and safety profiles of these compounds are lacking. COMRADE-C is the first head-to-head phase IIIb study to compare the efficacy and safety of the European labels of ranibizumab vs dexamethasone intravitreal implant, used according to the 2015 European Medicines Agency (EMA) labels, in patients with visual impairment due to macular edema secondary to CRVO, according to valid treatment recommendations.
Methods
Study Design
COMRADE-C was a 6-month, phase IIIb, multicenter, randomized double-masked controlled clinical trial that enrolled patients with visual impairment due to macular edema secondary to CRVO. Patients were enrolled from 66 sites across Germany, Great Britain, Poland, and Hungary. The study was conducted between August 2011 and January 2014 in accordance with the International Conference on Harmonization Harmonized Tripartite Guidelines for Good Clinical Practice, with applicable local regulations, and the Declaration of Helsinki. The study protocol was reviewed and approved by an independent ethics committee, an institutional review board, or a research ethics board for each contributing center. All patients provided written informed consent before entering the study. The COMRADE-C study is registered with clinicaltrials.gov under NCT01396083 .
Patients
The study population consisted of (1) male and female patients aged ≥18 years with visual impairment due to macular edema secondary to CRVO diagnosed ≤6 months before screening, and (2) patients with BCVA (study eye) of 20/40 to 20/400 (6/12 to 6/120 meters) using Early Treatment Diabetic Retinopathy Study (ETDRS)-like VA testing charts.
Key exclusion criteria were as follows: (1) a history of radial optic neurotomy or sheathotomy in the study eye, presence of either dry or wet age-related macular degeneration (AMD) in the study eye, ocular diseases (uveitis, neovascular glaucoma, diabetic retinopathy, diabetic maculopathy, or ocular ischemic syndrome) associated with increased intraocular VEGF levels, macular detachment/subretinal fluid attributable to causes other than BRVO, hypersensitivity to any of the study drugs or to drugs with similar chemical structures, or allergy to fluorescein; (2) central subfield retinal thickness (CSRT) of <250 μm in the study eye; (3) prior episode of retinal vein occlusion in the study eye; (4) anti-VEGF treatment in the study or the fellow eye 3 months before baseline; (5) panretinal scatter photocoagulation or sector laser photocoagulation within 3 months before baseline or anticipated within the 4 months following randomization; (6) intraocular corticosteroid use within 6 months before baseline; (7) IOP of ≥30 mm Hg or uncontrolled glaucoma; patients could be rescreened after 1 month if they had undergone treatment; (8) a history of cerebral vascular accident or myocardial infarction within 12 months prior to baseline; and (9) a history of pars plana vitrectomy.
Randomization and Treatment
A randomization list was produced by a validated system that randomly assigned the treatment arms to randomization numbers in the specified ratio. At enrollment, patients were randomly allocated in a 1:1 ratio to receive either the ranibizumab 0.5 mg intravitreal injections or dexamethasone intravitreal implant 0.7 mg. If both eyes were eligible, the eye with worse VA (assessed at visit 1) was selected, unless the investigator deemed that the other eye was more appropriate for study treatment. All patients received treatment in accordance with the European Union summary of product characteristics (SmPC) for ranibizumab or dexamethasone intravitreal implant. As mandated by the study protocol, no adjustments of the ranibizumab or dexamethasone intravitreal implant dosing regimen, or rescue therapy, were allowed.
Ranibizumab group
Patients received intravitreal ranibizumab 0.5 mg injections as per the 2011 European (EU) SmPC. They received a minimum of 3 consecutive monthly ranibizumab injections or injections until stable VA (no change in VA for 3 consecutive monthly assessments based on investigators’ judgment) was reached; thereafter, the patients were monitored monthly for VA. Monthly treatment was resumed if there was a loss of VA owing to disease activity, and it was continued until the VA was again stable for 3 consecutive months (implying a minimum of 2 injections during stable VA) ( Supplemental Figure 1 ; Supplemental Material available at AJO.com ).
Dexamethasone group
Patients received a single implant of sustained-release intravitreal dexamethasone intravitreal implant 0.7 mg at baseline according to the approved EMA label. Thereafter, they received monthly sham injections, according to the ranibizumab injection scheme, for the entire 6-month study period. The sham injections consisted of empty sterile syringes without needles and the injections were mimicked by applying pressure against the globe of the eye ( Supplemental Figure 1 ; Supplemental Material available at AJO.com ).
Masking and randomization
A minimum of 2 investigators were involved at each study site to fulfill the masking requirements. All ocular assessments were carried out by the evaluating physician, who was masked to the treatment assignment. The injecting physicians were unmasked and performed the study drug administration as per protocol; however, they were not involved in any other aspects of the study and were not allowed to communicate the details of the treatment to anyone. Decisions regarding retreatment were made by masked investigators.
Study Objectives
The primary objective was to compare the response to treatment with ranibizumab with that of dexamethasone intravitreal implant, with respect to the mean average change in BCVA from baseline to month 1 through month 6. The secondary and key exploratory objectives were to compare the efficacy, safety, and health-related quality of life (HRQoL) of ranibizumab with dexamethasone intravitreal implant for the following: (1) mean change in BCVA and CSRT over time from baseline to month 6; (2) proportion of patients with a BCVA gain or loss of ≥15/≥10/≥5 letters at month 6; (3) time taken to achieve a significant improvement in BCVA, defined as ≥15 letters at month 6; (4) number of injections during the study; (5) IOP increase from baseline to month 6; (6) safety over time; and (7) changes in HRQoL according to the National Eye Institute Visual Function Questionnaire (NEI VFQ-25), the Short Form Health Survey (SF-36), and the Euro Quality of Life (EQ-5D) questionnaire from baseline to month 6.
Efficacy and Safety Assessments
Best-corrected visual acuity
The BCVA of the study eye was assessed at every visit using ETDRS-like VA testing charts at a testing distance of 4 meters by a certified examiner. If the patient was unable to read at least 4 letters at 4 meters, the distance was reduced to 1 meter. The primary endpoint was the average of the changes in BCVA (letters) from baseline to any postbaseline visit until month 6. This endpoint provides a more robust estimate, as it accounts for both interpatient and intramonth variability in BCVA, as compared with mean change assessed at a single time point (month 6, end of the study).
Color fundus photography and fluorescein angiography
Color fundus photography in conjunction with fluorescein angiography (FA) was conducted at screening and at the end of the study. Color fundus photographs and FA images were independently reviewed by a central reading center (CRC; GRADE reading center, Bonn, Germany) to ensure standardized evaluation.
Optical coherence tomography
Optical coherence tomography (OCT; if available, spectral-domain OCT was used (n = 243); if not available (n = 11), time-domain OCT was used) was conducted at all the visits. Retinal thickness was assessed in terms of CSRT (by the investigator) and foveal center point thickness (FCPT) as assessed by the CRC, the latter to exclude possible measuring inaccuracies of different OCT. Here, FCPT was defined as the distance from the internal limiting membrane (ILM) to the bottom edge of the Bruch membrane at the fovea centralis.
Health-related quality of life
HRQoL was assessed using the vision-specific NEI VFQ-25 and the generic health assessment utility tool, SF-36, and the indirect utility questionnaire EQ-5D (0 = worst score; 100 = best score). All questionnaires were scored by patients at baseline and at month 6. Endpoints included absolute scores and absolute change from baseline. The results will be reported separately.
Treatment exposure
The number of injections (ranibizumab or dexamethasone intravitreal implant or sham) administered to each treatment group was evaluated over 6 months.
Safety assessments
The incidence of ocular and nonocular adverse events (AEs) and serious adverse events (SAEs), including their relationship to the study treatment and/or ocular injection procedure, was assessed during the 6-month study period. Safety was evaluated by slit-lamp examination and ophthalmoscopy, IOP measurements (≥10% increase in IOP at any postbaseline visit), changes in vital signs, and laboratory evaluations over the 6-month study period. AEs were summarized by the proportion of patients experiencing AEs based on the standardized Medical Dictionary for Regulatory Activities by system organ class and preferred term.
Study withdrawal and study drug discontinuation
Patients deemed to have an unsatisfactory therapeutic effect or recurrence prior to completion of the trial discontinued study treatment and received therapy as needed at the discretion of the investigator. Treatment failure was defined by the absence of visual improvement at visits 5, 6, and 7 compared to baseline BCVA at visit 1; reduction of central retinal thickness ≤ 10% (250 μm ≤ CRT baseline ≤ 500 μm) or 50 μm (CRT baseline ≥ 500 μm) compared to baseline CRT (CRT baseline) at visit 1; or decrease in BCVA ≥30 letters in the study eye compared with last assessment of BCVA prior to most recent treatment. Patients were also withdrawn from the study at any time if the investigator concluded that participation in the trial would put their safety at risk or withdrawal would be in the patient’s best interests.
Conditions requiring discontinuation from treatment included rhegmatogenous retinal detachment, development of macular hole stage III-IV, transient ischemic attack (TIA), or stroke. Criteria for study drug (ranibizumab and Ozurdex) interruption until the condition resolved or had been treated successfully included intraocular inflammation, elevated IOP in the study eye ≥30 mm Hg, occurrence of retinal break in the study eye, ocular or periocular infection, or intraocular surgery in the study eye within the previous 28 days.
Statistical Analysis
A sample size of 108 patients in each of the treatment groups was required to achieve 90% power to detect a significant difference on a 2-sided, 5% significance level, if the true difference was 6 letters with a common standard deviation (SD) of 13.5 letters. In order to compensate for the dropouts and other protocol violations, 120 patients were considered in each treatment group.
Primary analysis was performed on the full analysis set (FAS) that comprised all the patients who received at least 1 application of study treatment and had at least 1 postbaseline assessment for BCVA. The primary analysis was performed using the last observation carried forward (LOCF) approach and analysis of covariance model, with the average change in BCVA (letters) from month 1 through month 6 as a dependent variable; and with the factors center, treatment, and covariate baseline BCVA.
Raw means, SD, and least-square (LS-) means with corresponding 95% confidence intervals and P values were calculated as point estimates for the treatment contrasts. The null hypothesis stated that “no difference in the change of BCVA averaged over all postbaseline visits.”
Sensitivity analysis of the primary analysis was performed within the FAS by comparing the primary analysis (based on LOCF) with the as-documented approach that used the observed values only without imputation for missing values. Further sensitivity analysis was performed using a mixed model for repeated measures (MMRM) as well as repeating the primary analysis on the per-protocol set (PPS, as observed). The time to BCVA gain of ≥15 letters was analyzed using the Kaplan-Meier method.
The safety analyses were conducted on the safety set that comprised all patients who had received at least 1 application of the study treatment and had at least 1 postbaseline safety assessment. AEs were summarized by presenting the number and percentage of patients with any ocular/nonocular AEs. Deaths, SAEs, and AEs leading to study discontinuation for study treatment were listed separately and were summarized by the primary system organ class and preferred term.
Results
Patient Demographics and Baseline Characteristics
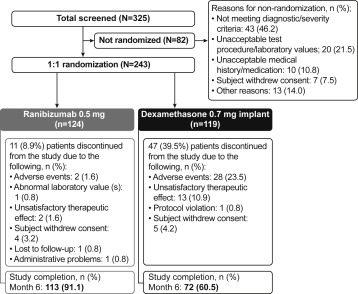
Of 325 patients screened, 243 were randomly allocated to receive 1 of the drugs: 124 to ranibizumab and 119 to dexamethasone intravitreal implant. Overall, 113 patients (113/124; 91.1%) in the ranibizumab group and 72 patients (72/119; 60.5%) in the dexamethasone intravitreal implant group completed the 6-month study. The main reasons for discontinuation in the ranibizumab and dexamethasone intravitreal implant groups were AEs (1.6% and 23.5%, respectively), unsatisfactory therapeutic effect (1.6% and 10.9%, respectively), and withdrawal of consent (3.2% and 4.2%, respectively) ( Figure 1 ). Efficacy and safety analyses were conducted on the FAS and safety set, respectively, with each set comprising 124 patients from the ranibizumab group and 119 patients from the dexamethasone intravitreal implant group.
Overall, patient demographics and baseline ocular characteristics were similar between the 2 treatment groups, as shown in the Table . At baseline, 93.5% of patients in the ranibizumab group and 95% of patients in the dexamethasone intravitreal implant group had active concomitant medical conditions, the most common being hypertension (62.1% and 65.5%, respectively), hypercholesterolemia (23.4% and 21%, respectively), and hypothyroidism (14.5% and 16.0%, respectively). It is important to note that 18.2% of patients in the ranibizumab group and 16.1% of patients in the dexamethasone intravitreal implant group showed retinal ischemia at baseline (defined as enlargement of the foveal avascular zone) and 6.5% in the ranibizumab group and 4.2% in the dexamethasone intravitreal implant group were pseudophakic.
| Characteristics | Ranibizumab 0.5 mg (n = 124) | Dexamethasone 0.7-mg Implant (n = 119) | Total (N = 243) |
|---|---|---|---|
| Mean age (SD), y | 65.3 (11.4) | 66.9 (12.4) | 66.1 (11.9) |
| Sex, n (%) | |||
| Male | 72 (58.1) | 73 (61.3) | 145 (59.7) |
| Female | 52 (41.9) | 46 (38.7) | 98 (40.3) |
| Race, n (%) | |||
| White | 119 (96.0) | 118 (99.2) | 237 (97.5) |
| Asian | 3 (2.4) | 0 (0.0) | 3 (1.2) |
| Black | 1 (0.8) | 0 (0.0) | 1 (0.4) |
| Other | 1 (0.8) | 1 (0.8) | 2 (0.8) |
| Mean time (SD) since diagnosis of CRVO, d b | 44 (40) | 45 (56) | 45 (48) |
| Mean time (SD) since diagnosis of macular edema, days | 38 (36) | 42 (56) | 40 (47) |
| Mean baseline BCVA (SD), letters | 51.7 (16.5) | 51.5 (15.6) | 51.6 (16.1) |
| Baseline CSRT | |||
| N | 122 | 119 | 241 |
| Mean (SD), μm | 723.8 (245.9) | 705.2 (231.1) | 714.6 (238.4) |
| Baseline FCPT | |||
| N | 117 | 116 | 233 |
| Mean (SD), μm | 736.9 (280.5) | 778.7 (266.9) | 757.7 (274.0) |
a Comprises all randomized patients who received at least 1 application of the study treatment and had at least 1 postbaseline assessment for BCVA.
b Mean time in days from first diagnosis of CRVO to baseline visit in clinical trial.
Protocol deviation
Noticeable differences for major protocol deviations were observed between the 2 treatment groups. Differences were mainly linked to the following deviations (which occurred less frequently with ranibizumab than with dexamethasone intravitreal implant): dropouts (10 [8.1%] vs 47 [39.5%]), missing study injection (6 [4.8%] vs 16 [13.4%]), forbidden concomitant medication–anti-VEGF agents (1 [0.8%] vs 12 [10.1%]), and unmasking (1 [0.8%] vs 7 [5.9%]). Other major protocol deviations were documented in <3% of the overall patient population. A greater number of patients dropped out from the dexamethasone intravitreal implant group than from the ranibizumab group because of AEs or unsatisfactory treatment. AEs that led to study discontinuation with dexamethasone intravitreal implant were mainly related to eye disorders such as reduced VA and macular edema, mainly because of treatment failure at visits 5, 6, and 7 ( Supplemental Table 1 ; Supplemental Material available at AJO.com ).
Efficacy
Best-corrected visual acuity
At months 1 and 2 there was no difference in mean (± SD) change in BCVA from baseline between ranibizumab and dexamethasone intravitreal implant, suggesting similar initial efficacy. At month 3, improvement in mean (± SD) BCVA was significantly higher with ranibizumab compared to dexamethasone intravitreal implant (raw mean ± SD: 16.0 ± 13.4 vs 7 ± 18.2 letters). This difference between the treatment groups further increased until month 6 (raw mean ± SD: +16.9 ± 13.6 letters for ranibizumab and −0.7 ± 22.5 letters for dexamethasone intravitreal implant; Figure 2 ). The LS-means [95% CI] at month 6 were 14.78 [11.24–18.32] letters with ranibizumab and −3.17 [−6.8 to 0.46] letters with dexamethasone intravitreal implant. The difference between the treatment groups was 17.96 (95% CI: [13.37–22.54]) letters. Sensitivity analyses revealed consistent results for the MRMM, as well as the as-observed analyses compared to the primary LOCF approach, showing robustness of results regardless of the missing values. However, sensitivity analysis on the PPS (as observed) yielded a lower difference in the mean average change in BCVA as compared to the primary result, which may in part result from the low sample size in the PPS ( P = .0904).