Mucous membrane pemphigoid (MMP) is a spectrum of acquired, autoimmune, subepithelial blistering diseases that primarily affect mucous membranes but may involve the skin. Morbidity is associated with scar formation and may be especially severe when mucosal surfaces such as the conjunctivae, larynx, esophagus, or urethra are involved. Bilateral, albeit frequently asymmetrical, progressive subconjunctival scar formation is the key ocular feature in ocular MMP, otherwise known as ocular cicatricial pemphigoid (OCP) (
1,
2,
3,
4). Loss of vision or even loss of the eye may result from corneal involvement.
Epidemiology
OCP is uncommon. Estimates suggest that 1 in 8,000 to 46,000 ophthalmic patients, depending on the series, have OCP (
5,
6,
7). The disease may be more common than is recognized, however, given the exclusion from these numbers of patients presenting with predominantly skin or nonocular mucosal involvement. Also, the diagnosis of this disorder in its early stages is difficult, and most cases of early disease may be uncounted in the epidemiologic estimates. MMP has no geographic or racial predilection, but a genetic susceptibility has been suggested (
8,
9). It is a disease of relatively older populations, with a peak of presentation between the sixth and seventh decades (range 43 to 88 years) (
10). Although considered to be a disease of the elderly, in many patients, because of the subtle nature of the subepithelial fibrosis in the earliest stages of the disease, it may have begun in the fourth or fifth decade of the life. Most series report a female predilection, with a female to male ratio of approximately 2:1 (
1,
8).
Clinical Features
MMP is a group of chronic inflammatory, blistering diseases predominantly affecting any or all of the mucous membranes, with or without clinically apparent scar formation. OCP is one subset of this group.
Ocular Involvement
At its onset and in the early stages, OCP is usually clinically indistinguishable from any other nonspecific chronic conjunctivitis. Patients frequently complain of conjunctival irritation, burning, tearing, hyperemia, and discharge. The earliest ocular manifestation of OCP is an intermittent, unilateral chronic papillary conjunctivitis, which eventually becomes bilateral. Foreign-body sensation and photophobia can occur, as a consequence of breakdown of the corneal epithelium (
11,
12,
13).
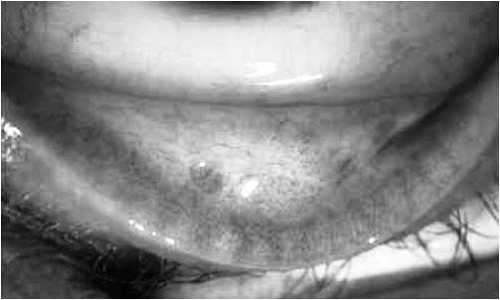
Later in the course of the disease, conjunctival bullae can form. These are rarely seen by the ophthalmologist, as they evolve quickly into ulcerous lesions, conjunctival thickening, and scarring. The earliest sign of conjunctival scarring is formation of lacy white lines of subepithelial fibrosis, commonly perivascular in localization and appearing first in the inferior fornix (
1,
12,
14) (
Fig. 20-1). Another early clinical sign is involvement of the canthal structures, leading to shallow canthal recesses (
Fig. 20-2). Involvement of the medial canthus can lead to a loss of architecture, with flattening and obliteration of the normal conjunctival folds, plica, and caruncula (
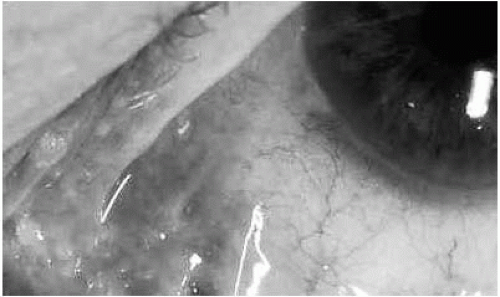
15). As the disease advances, progression of the subepithelial fibrosis results in “shrinkage” of the fornices and then formation of symblephara, fibrotic bands between the palpebral and bulbar conjunctiva.
Early symblephara are best demonstrated by external examination with a penlight, retracting the lower eyelid downward while the patient looks upward.
During the chronic stage, the disease manifests as a chronic conjunctivitis with cicatrix formation. Fibrosis beneath the conjunctival epithelium in the upper tarsal conjunctiva and superior fornix can cause obliteration of the ducts of the lacrimal and accessory lacrimal glands, resulting in keratoconjunctivitis sicca (
13). Excessive and progressive conjunctival shrinkage later in the disease course alters the architecture of the lids and causes lagophthalmos and exposure, or entropion. Severe prolonged conjunctival cicatrization, in the absence of effective therapy, may cause ankyloblepharon, fusion of the lower eyelid to the bulbar conjunctiva, resulting in a restriction of ocular motility (
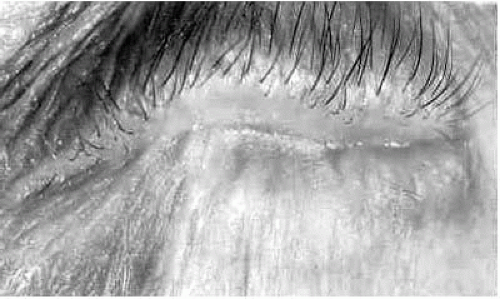
1,
13,
14,
15,
16) (
Fig. 20-3). Alterations in the orientation of eyelash follicles as a consequence of the scarring process can produce trichiasis and distichiasis, and squamous metaplasia with keratinization of the eyelid margins may occur. All these factors damage the corneal epithelium.
The advanced stages of the disease include blinding keratopathy, with corneal neovascularization, pseudopterygium formation, and progressive thinning and perforation. Secondary bacterial infection and ulcers may be associated with the compromised corneal epithelium. Use of topical corticosteroids, use of bandage contact lenses, chronic irritation, and epithelial defects resulting from trichiasis, meibomitis, and lagophthalmos may predispose to development of bacterial superinfections.
OCP-induced alterations in aqueous outflow may also predispose to development of glaucoma. Indeed, in a series of 111 patients, 29 (26%) had glaucoma (
17). Interestingly, 27 of these patients had a history of glaucoma for a mean of 11.3 years before the diagnosis of OCP. Most had advanced glaucoma, with optic nerve damage and visual field loss.
Mucous Membrane Involvement
MMP also involves mucous membranes, other than conjunctiva. Oral lesions have been found to be the most common manifestation, occurring in 91% of patients in a series of dermatologic patients with MMP (
18). In the same series, the conjunctival lesions were present in 66% of the patients.
In ophthalmic series (all patients having conjunctival involvement), 15% to 50% also had oral mucosal lesions (
1,
10,
19).
The most common finding in the mouth is desquamative gingivitis, which may be patchy or diffuse, with redness, easy bleeding, and ulcerations (
14,
20,
21). Another form of oral mucosal involvement is vesicle and bulla formations of the oral mucosa that develop rapidly, remain intact for a few days, and then rupture (
22). Involvement of the larynx, trachea, or esophagus also occurs. Esophageal involvement is not uncommon and has been reported in as many as 27% of patients (
23); it may lead to heartburn, dysphagia, and fatal aspirations (
24). The laryngeal and tracheal involvement with stenosis may cause difficulty in breathing, intermittent hoarseness, or dysphonia, and this requires endoscopic evaluation (
25,
26). Because many patients first present with ocular symptoms, ophthalmologists must inquire about signs of potential life-threatening esophageal and laryngeal involvement, such as hoarseness and difficulty in breathing or swallowing. Scarring and stenosis may lead to fatalities and thus warrant special attention and referral for evaluation.
Cutaneous Involvement
Skin lesions occur in 9% to 24% of patients with MMP, depending on the series (
1,
4,
18). It is less frequent than mucous membrane involvement and can be of two types: (a) recurrent vesiculobullous, nonscarring eruptions that involve the extremities and inguinal area (similar to but smaller than those seen in bullous pemphigoid); or (b) localized erythematous plaques with overlying vesicles and bullae of the scalp and face that evolve into pruritic blisters that rupture and leave scars (Brusting-Perry dermatitis) (
13,
25).
Associated Medical Conditions
OCP can be associated with other “autoimmune” disorders. An association with rheumatoid arthritis has been noted (
27) and confirmed by others (
1,
28). In the largest series of 130 patients with OCP, 23 patients had an associated medical condition (
1). Nineteen of the 130 (15%) had rheumatoid arthritis. Also, in the same series, three patients had systemic lupus erythematosus, and one had multiple autoimmune phenomena. Wegener’s granulomatosis has also been reported to occur simultaneously with MMP (
29).
Pathogenesis and Immunology
Mucous membrane pemphigoid is an autoimmune disease characterized by the presence of autoantibodies, most commonly immunoglobulin G (IgG), that bind to autoantigens located in the conjunctival epithelial basement membrane zone (BMZ), and of components of both classical and alternative complement pathways. A T-cell dysregulation, abnormal serum levels of cytokines such as interleukins (IL)-1, IL-5, IL-6, and tumor necrosis factor (TNF)-α and -β can be detected (
30).
There is a genetic predisposition to OCP with an increased incidence of human leukocyte antigen (HLA)-DQB1
*0301 gene (DQw7) (
9). HLA-DR2 (
31) and HLA-DR4 antigens (
32) have also been implicated. HLA-DQ molecules on the surface of antigen-presenting cells function immunologically by presenting antigen to T cells. The DQB1
*0301 gene may have a role in T-cell recognition of basement membrane antigens, resulting in production of anti-BMZ IgG autoantibodies (
33).
By virtue of the genetic susceptibility, some triggering agent(s) during the patient’s life can cause development of autoantibodies to the target autoantigens at the BMZ. Some identified environmental triggers include systemic practolol therapy (
34) and ocular exposure to epinephrine (
35), idoxuridine (
36), phospholine iodide (
37), and Humorsol (
38). The autoantigens located in the BMZ include the β4 protein of the α6β4 integrin as a highly targeted autoantigen (
39,
40) and epiligrin (α3 subunit of laminin 5) (
41).
Binding of the autoantibody to the target autoantigen at the epithelial BMZ is followed by activation of the complement cascade, with deposition of the complement products and inflammatory cells predominantly at the level of the basal epithelial hemidesmosome and the lamina lucida of the basement membrane (
14). Lymphocytes, macrophages, dendritic cells, plasma cells, neutrophils, and mast cells are recruited to the subepithelial tissues in different compositions depending on the clinical activity of the disease. OCP is not simply an antibody-mediated disease, but rather involves T-cell dysregulation as well (
42).
There is evidence that soluble factors, especially fibrogenic cytokines secreted by inflammatory cells and fibroblasts, play an important role in the immunopathogenesis of acute MMP by remodeling the matrix in the conjunctival stroma, possibly by regulating the altered metabolism of matrix proteins (
43). Studies on cytokine profiles have shown an increased expression of transforming growth factor (TGF)-β (
43,
44,
45) and IL-5 (
30), decreased levels of serum IL-6 (
45), and elevated
levels of serum IL-1 and IL-1 components (
46) in patients with active OCP.
The absence of fibrogenic cytokines in chronic progressive OCP provides support for the idea that fibroblasts in the conjunctiva of patients with OCP may remain functionally and morphologically abnormal after withdrawal of cytokine influences (
43).
Histopathology
The characteristic conjunctival scarring in OCP is a consequence of subepithelial fibrosis. Accumulating evidence indicates that the subepithelial fibrosis is mainly caused by macrophages and by conjunctival fibroblasts (
1). The number of macrophages within the conjunctival subepithelial tissue is significantly increased in OCP and reflects the activity of the disease. One study has demonstrated a relationship in OCP between local proliferation of macrophages and increased expression of macrophage-colony-stimulating factor (m-CSF), mainly by conjunctival fibroblasts and infiltrating inflammatory cells (
47). Macrophages, by producing fibrogenic cytokines such as TGF-β, platelet-derived growth factor, and basic fibroblast growth factor, stimulate the proliferation and migration of the fibroblasts—the cellular elements responsible for fibrosis (
12). The conjunctival fibroblasts from patients with OCP are abnormally hyperproliferative, with a decreased doubling time in tissue cultures when compared with normal cells (
48). These activated fibroblasts produce an abnormal new extracellular matrix and collagen, mostly type III (
49). Thus the scarring that characterizes MMP appears to be due to excessive fibroblast proliferation and abnormal collagen synthesis.
The conjunctival epithelium in OCP shows squamous metaplasia with parakeratosis and keratinization (
50), associated with a decreased number or absence of mucin-producing goblet cells (
51). Destruction of the goblet cells and obliteration of the lacrimal gland ductules cause severe dry eye. The inflammatory cells in the conjunctiva of OCP patients are predominantly mononuclear cells, mainly T lymphocytes. There is a threefold increase in T lymphocytes within the epithelium and a 20-fold increase within the substantia propria of bulbar conjunctival tissue of patients with OCP (
42). These T cells are activated, as evidenced by the expression of IL-2 receptors on their cell surfaces. The dendritic cells are also increased, 25-fold, over normal levels. The number of macrophages and neutrophils was also found to be increased, particularly in the acute stages of the disease, and correlate with the clinical activity of the disease (
44). Interestingly, the infiltrations in subacute and chronic OCP differ greatly. The number of mononuclear cells, neutrophils, CD45
+ cells, and CD3
+ cells, as well as HLA-DR expression, were significantly higher in the subacute than in the chronic disease, with some eosinophil granule proteins such as eosinophil-derived cationic protein and major basic protein in the epithelium and substantia propria of the conjunctiva in the subacute form (
52). A study of the mast cell subsets in the conjunctivae in OCP revealed a predominance of connective tissue mast cells (
53).
Diagnosis
Unfortunately, the diagnosis of OCP is frequently delayed because of the nonspecificity of its early symptoms and the difficulty of recognizing the early stages. Patients are usually diagnosed with MMP when they already have advanced signs of conjunctival cicatrization and ocular surface disturbances. To reduce the delay in diagnosis, a careful ocular and systemic examination must be performed for any patient with conjunctival scarring. The clinician should inquire about the presence of oral lesions, past and present oral and topical medications, any history of previous ocular infection, drug reaction, trauma, chemical burns, ionizing radiation, atopy, and conjunctival surgery. Facial skin, scalp, nasal and oral mucosa, finger and wrist joints, nail beds, and periungual folds should be closely examined for any associated systemic or iatrogenic disorders. Because MMP may lead to blindness, it is of critical importance to make the diagnosis as early as possible to allow early treatment.
Conjunctival biopsy facilitates the early diagnosis of MMP. A lower bulbar conjunctival specimen is harvested from the actively inflamed eye and divided into three equal samples for light microscopic, electron microscopic, and immunohistochemical studies. Linear immunoreactant (immunoglobulin and/or complement) deposition at the BMZ confirms the diagnosis (
54,
55). Other disorders such as bullous pemphigoid, epidermolysis bullosa acquisita, and paraneoplastic and drug-induced cicatrizations may produce a similar pattern. Also, absence of immunoreactant does not exclude MMP, and regrettably, false-negative biopsy occurs in a significant proportion of patients (
3,
12). Immunohistochemical analysis of biopsied conjunctiva by direct immunofluorescence yields a positivity rate of 50% to 60% (
55,
56). Routine use of the immunoperoxidase technique in immunofluorescent-negative biopsies, allied with appropriate harvesting and handling of conjunctiva, increases the diagnostic yield to more than 80% (
56). Especially in cases of multiple negative biopsies, other specimens submitted for light microscopic and electron microscopic studies should be carefully examined to look for other possible causes of cicatrizing conjunctivitis.
The early diagnosis of MMP is challenging yet essential, because the ocular manifestations may be the first clinical presentation of this potentially fatal disease.
Differential Diagnosis
MMP must be differentiated from a variety of disorders that can cause cicatrizing conjunctivitis (
Table 20-1). The chronic progressive course of conjunctival cicatrization, the sine qua non of a diagnosis of MMP, can also be seen in linear IgA disease, bullous pemphigoid, epidermolysis bullosa acquisita, and drug-induced conjunctival cicatrization, in a relatively milder form. As in MMP, direct immunofluorescence shows linear immune deposits at the BMZ in linear IgA disease, epidermolysis bullosa acquisita, and bullous pemphigoid. In MMP, epidermolysis bullosa acquisita, and bullous pemphigoid, the deposits mainly consist of IgG and C3, but in linear IgA disease they are mainly IgA (
59). Circulating autoantibodies may be found in all these disorders, but are observed with higher frequency and higher titers in MMP. The morphology and distribution of eruptions may help in the differential diagnosis of these conditions.
Clinically, involvement of canthal structures and upper tarsal conjunctiva is an important early clinical sign of MMP; in contrast, the conjunctival cicatrization due to topical or systemic medications involves mainly the lower fornix (
15,
60).
Cicatrizing conjunctivitis resulting from chemical burns, radiation exposure, and membranous conjunctivitis is generally nonprogressive, unlike that in MMP (
11). Stevens-Johnson syndrome should also be considered in the differential diagnosis. However, the initial acute illness, typical skin lesions, and history of exposure to a known trigger easily differentiate this condition from MMP.
Treatment
The natural history of MMP is such that, if untreated, chronic gradual progression of the inflammation leads to bilateral blindness. Early diagnosis and therapy can prevent ocular complications, but advanced stages of the disease often result in irreversible visual loss despite the institution of therapy. The objectives of management of MMP involves control of the systemic immunodysregulation, inhibition of the progressive conjunctival scarring, and maintenance of a normal relation between the eyelids and the ocular surface to prevent ocular surface disturbances. Because MMP is a systemic disease, no topical therapy is effective in controlling progression of the ocular inflammation and the scarring process. Failure of topical and subconjunctival treatment with corticosteroids, cyclosporine, mitomycin-C, and retinoids has been well demonstrated (
61). Once the diagnosis is established, long-term systemic therapy must be instituted as soon as possible.
Dermatologists evaluated the efficacy of systemic corticosteroids in the early 1970s (
62). Although high-dose oral prednisone was effective in preventing progression of the mucosal shrinkage, many of the patients developed unacceptable steroid-induced complications because of the high dose required. Azathioprine was then shown to be beneficial as a nonsteroidal immunomodulatory therapy (
63).
Soon after came a report of the cure of a patient with systemic cyclophosphamide therapy (
64). In the early 1980s the results of the first large ophthalmic case series demonstrating the efficacy of systemic immunosuppression were published, reporting cessation of the inflammation and cicatrization with azathioprine or cyclophosphamide combined with systemic steroid therapy (
57). Another large study followed (
21).
Formulation of a standard treatment protocol for MMP is very difficult. The therapeutic approach should take into consideration the site involved, the severity of involvement, and the rapidity of progression. The importance of considering the specific site is based on the observations that ocular involvement can lead to blindness, tracheal and laryngeal involvement can lead to airway obstruction, and genital involvement can lead to urinary and sexual dysfunction.
Diaminodiphenylsulfone (dapsone) is the usual initial chemotherapeutic agent for patients with mild to moderate inflammation and slow progression of the disease (
65,
66). Dapsone should not be given to patients with a history of sulfa allergy or glucose-6-phosphate dehydrogenase deficiency. It has potentially severe adverse effects such as hemolytic anemia, agranulocytosis, aplastic anemia, hepatitis, and peripheral neuropathy. A complete blood count, liver function tests, and glucose-6-phosphate dehydrogenase activity should be evaluated before initiating therapy. The usual initial therapeutic dose of dapsone is 25 mg twice daily. If the drug is tolerated well and has no side effects, the dose can be increased to 50 mg twice daily if necessary. The highest dose employed is 150 mg/day. If the patient has taken the drug for 12 weeks with no response, advancement to more conventional systemic immunosuppression with methotrexate (7.5-15 mg once weekly), or daily azathioprine (2-3 mg/kg), or daily mycophenolate mofetil (1-2 g/day) follows (
49). In severe or rapidly progressive forms, the first-line treatment of choice is systemic prednisone (1 mg/kg/day) combined with cyclophosphamide (2 mg/kg/day). To avoid long-term complications, steroids should never be used as sole agents in treating MMP, and tapering of steroids should begin within 6 weeks of initiating treatment. Analyses of success rates with the recommended systemic immunosuppression approach show that approximately 6% to 10% of patients continue to experience progression of inflammation with irreversible damage to the ocular surface, despite the best treatment efforts (
21,
67).
In progressive cases, intravenous immunoglobulin treatment can be used as an alternative to the systemic immunosuppressive agents (
68,
69). Intravenous infusions of pooled human Ig, 2 to 3 g/kg/cycle divided over 3 days and repeated every 2 to 6 weeks, is the usual regimen. Intravenous Ig exhibits a number of immunomodulatory properties that are mediated by the Fc portion of IgG and by the spectrum of variable regions contained in the immunoglobulin preparations (
70). Although the major component of intravenous Ig is IgG, other minor components such as solubilized lymphocyte surface molecules also exhibit immunoregulatory effects on T- and B-cell immune responses. The lack of significant side effects makes intravenous Ig a valuable therapeutic agent.
Daclizumab is a humanized IgG monoclonal antibody produced by recombinant DNA technology that specifically binds CD25 of the human IL-2 receptor expressed on activated T lymphocytes. The medication is composed of 90% human and 10% murine antibody sequences that function as an IL-2 receptor antagonist for inhibiting IL-2 binding, thus selectively inhibiting activated but not resting T cells (
71). Daclizumab is administered intravenously at a dose of 1 mg/kg per treatment, with treatment every 2 weeks during the first 12 weeks of therapy, every 3 weeks until week 24 of therapy, then every 4 weeks until week 52. In total, the patient receives 18 doses of the medication during an entire year. Preliminary experience using daclizumab in the treatment of resistant cases shows promise (
72).
The treatment recommendations presented here are modified from the consensus statement of the First International Consensus on Mucous Membrane Pemphigoid, published in 2002 (
73).
Prolonged periods of remission without therapy can be maintained in approximately one third of patients with MMP following systemic immunosuppressive treatment. Follow-up must be continued for life, as relapses occur in 22% of those in remission off therapy (
74). It is also essential to involve an expert chemotherapist for the long-term care of patients with MMP, to monitor for potential toxicity and side effects of the medications.
Rehabilitation
Management of the ocular surface problems in MMP patients is extremely important. Severe abnormalities of the tear film with dry eye can be treated with artificial tears and lubricating ointments, preferably preservative-free, or with punctal occlusion if scarring has not already occluded the puncta. In the presence of trichiasis and lid abnormalities, therapeutic contact lenses can be used in patients with sufficient forniceal depth, to prevent the cornea from epithelial breakdown. Fluid-ventilated, gas-permeable scleral lenses are a valuable tool in the management of severe ocular surface problems. In addition to enhancing vision, they have the potential to reduce greatly the disabling ocular pain and photophobia and the rate of persistent/recurrent corneal epithelial defects. The therapeutic benefits of these lenses result from the oxygenated aqueous environment created over the corneal epithelium. The oxygenated precorneal fluid compartment maintained at neutral pressure protects the epithelial surface from the desiccating effects of exposure to air and the friction generated by blinking, and avoids the shearing forces generated during the blink-induced movement of soft lenses (
75).
Keratinization of the eyelids and conjunctiva stimulates the colonization of bacterial pathogens and places MMP patients at significant risk for ocular surface breakdown and bacterial blepharoconjunctivitis and keratitis. In more than 80% of patients, the eyelids or conjunctiva have colonies of potential pathogens (
4), so cultures should be taken frequently and if positive, the patient should be treated with topical antibiotics.
Surgery or any mechanical manipulation of the eyelids or conjunctiva should be strictly avoided during active stages of the inflammation, because the trauma of the surgery can trigger further conjunctival inflammation with resultant additional conjunctival scarring (
1,
76). All surgical procedures must be delayed until the conjunctival inflammation is completely controlled. After the eye becomes quiescent, procedures involving manipulation of the eyelids and conjunctiva or cataract surgery can be performed while the patient is taking immunosuppressive therapy. Penetrating keratoplasty may be performed to restore sight in patients with corneal pathology, after controlling the primary immunologic process and aggressive treatment of the mechanical factors damaging the ocular surface, such as anatomic lid problems from scarring, conjunctival and lid margin keratinization, and trichiasis, provided that the patient does not have significant dry eye. The success of penetrating keratoplasty in these patients is generally limited because of dryness and impairment of eyelid functions (
77). Limbal stem cell transplantation can be performed prior to or in combination with penetrating keratoplasty to improve outcomes, again provided that the effort is not sabotaged by a hostile environment such as dry eye, lagophthalmos, distichiasis (
78). However, patients with underlying immunologically mediated diseases generally have lower success rates with these procedures than do patients with noninflammatory ocular surface diseases (
79).
In patients with the final stages of MMP, keratoprosthesis is the only viable option to restore sight. Although the recent advances aimed at preventing and treating complications after keratoprosthesis surgery have improved prognosis, the potential complications are considerable: retroprosthetic membrane, glaucoma, endophthalmitis, extrusion of the prosthesis, and retinal detachment (
80). Dohlman’s success rate for achieving 20/200 or better vision in patients with OCP employing his keratoprosthesis is 33% after 2 years. However, at 5 years none of the seven patients retained vision better than 20/200 (
80).