This chapter addresses aspects of behavior disorders caused by damage to the visual cortex and white matter connections. These conditions are often referred to as “central disorders of vision,” “cerebral disorders of vision,” or “higher disorders of vision.” The understanding of these disorders continues to improve with the development of new techniques for measuring visual dysfunction, imaging the behaving brain, rehabilitating visual dysfunction in patients with brain damage, and even creating visual sensations in the blind with prostheses to stimulate visual cortex.
Vision was once thought to be primarily a serial (or hierarchic) process in which visual signals were altered or enhanced at successive way stations from retina to brain until an image of the physical world somehow emerged at the level of conscious experience. It has subsequently become clear that serial processing is only one of several mechanisms used by the brain to process visual signals. The primate visual system also uses parallel processing, beginning in the retina. Different types of retinal ganglion cells are specialized to transduce different types of physical signals and give rise to different channels. There is cross talk between these channels at several levels from the retina to the cortex, and there also are feedforward and feedback connections between early and late stages of the visual sensory system. Instead of just serial processing, visual functions are explained in terms of multiple interactions among specialized brain regions in the same hemisphere and across the corpus callosum. Central disorders of vision thus can be interpreted as a consequence of disturbing the processing in different sectors or pathways in a complex interconnecting network.
Segregation of Visual Inputs
The functional segregation of visual inputs in the primate visual system is well documented (Figs. 12.38 and 13.1). Retinal information is communicated to cortical neurons through a set of pathways that appear specialized to convey a particular class of visual information. For example, the parvocellular or P-pathway, named for its connections to simian striate cortex (area V1) via parvocellular layers 3 to 6 of the lateral geniculate body (LGB), is characterized by color opponency and slow-conducting axons that convey sustained signals. This pathway has strong projections to secondary areas such as V4 and inferior temporal (IT) cortex, located in the inferior occipital lobe and adjacent temporo-occipital regions. These regions, along the ventral or temporal cortical pathway (the “what” pathway), are presumed to play a role in the perception of color, luminance, stereopsis, and pattern recognition. In contrast, the magnocellular or M-pathway is characterized by large, fast-conducting axons that convey information about more transient visual signals. This pathway connects to areas in visual association cortex, including the middle temporal (MT) and medial superior temporal (MST) areas. These regions, located along the dorsal or parietal cortical pathway (the “where” pathway), are thought to
analyze the spatial location and movement of objects in the panorama (Fig. 13.1).
analyze the spatial location and movement of objects in the panorama (Fig. 13.1).
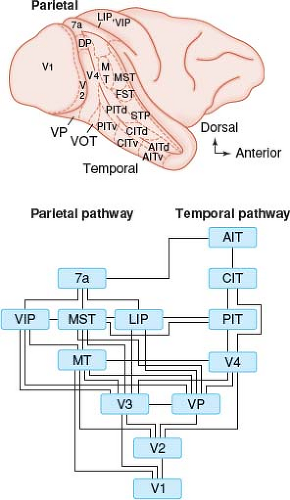 Figure 13.1 Parallel pathways for visual function in the cerebral cortex of the nonhuman primate. Borders between visual areas are indicated by fine dashed lines. The superior temporal sulcus has been opened to show areas that are normally out of sight. The medial hemisphere is not shown. The lower panel shows a flow diagram from area V1 (primary visual cortex) to the parietal pathway that includes area MT and the temporal pathway that includes area V4. The visual information encoded by neurons varies greatly among these different areas. The human brain is likely to respect a similar organization. 7a, Brodmann area 7a. AIT, anterior inferotemporal area (d, dorsal subdivision; v, ventral subdivision). CIT, central inferotemporal area (d, dorsal subdivision; v, ventral subdivision). DP, dorsal parietal area. FST, fundus of the superior temporal area. LIP, lateral intraparietal area. MST, medial superior temporal area. MT, middle temporal area. PIT, posterior inferotemporal area (d, dorsal subdivision; v, ventral subdivision). STP, superior temporal polysensory area. V1, visual area 1 (striate cortex). V2, visual area 2. V4, visual area 4. VIP, ventral intraparietal area. VOT, ventral occipitotemporal area. VP, ventral posterior area. (Reprinted with permission from Maunsell JHR. The brain’s visual world: Representation of visual targets in cerebral cortex. Science. 1995;270:765–769.) |
Analyses of data from a large number of patients with focal lesions of the visual cortex and its connections support the general concept of separate dorsal and ventral processing pathways as a framework for interpreting human clinical disorders. For example, damage to the inferior visual cortex and adjoining temporal regions impairs pattern recognition and learning, producing agnosia for objects and faces (prosopagnosia) or inability to read despite previous literacy (alexia). It can also reduce color perception in the contralateral field—cerebral achromatopsia. By contrast, damage to the superior visual cortex and the adjacent parietal cortex produces disorders of spatial-temporal analysis—inability to judge location, distance, orientation, size, or motion of objects, as well as marked disturbances of visually guided eye and hand control. Balint syndrome is a striking example.
Each of these major divisions probably has functional subdivisions. Furthermore, although the dorsal and ventral visual cortices are associated with different, behaviorally separable functions, there is growing evidence that these two major divisions also have overlapping functions.
Blindsight and Residual Vision
Some patients with lesions of area V1 causing a homonymous field defect perform better than chance on simple forced-choice detection tasks or on localization tasks measuring the accuracy of finger pointing or eye movements toward targets presented in the defective field of vision. Some of these patients deny any conscious experience of the objects they reportedly localize or detect and are thus said to have blindsight. This phenomenon is distinct from residual vision that may arise from a small population of spared neurons in V1, and precise anatomic analysis and clinical assessment are needed to ensure that this category of poor visual function is not mistaken for blindsight.
The dominant hypothesis in blindsight research is that the phenomenon represents residual function in a visual pathway parallel to the retino-geniculo-calcarine system. The initial candidate was an alternative pathway involving the superior colliculus, but more recent data implicate areas V5/MT. However, it has been difficult to completely exclude a contribution from residual activity of a small population of neurons in V1. In humans, a complete lesion is nearly impossible to confirm antemortem. Thus, research in nonhuman primates using fMRI before and after a permanent and complete surgical lesion of V1 has been conducted, and results indicate persistent albeit weak activation of V4 with presentation of stimuli in the lesioned hemifield. Further experiments are needed to confirm and expand these findings.
There are also explanations of blindsight that do not invoke parallel pathways. Blindsight may simply reflect a testing phenomenon known as criterion shift
in which patients tend to use fairly conservative criteria when asked to respond Yes or No during a detection task but use more relaxed criteria when forced to choose an alternative (i.e., guess) in a discriminatory paradigm. Furthermore, studies that attempt to determine if hemianopic patients have blindsight must eliminate all potential testing artifacts, including inadequate fixation, light scatter, nonvisual cues, and nonrandom presentation of targets.
in which patients tend to use fairly conservative criteria when asked to respond Yes or No during a detection task but use more relaxed criteria when forced to choose an alternative (i.e., guess) in a discriminatory paradigm. Furthermore, studies that attempt to determine if hemianopic patients have blindsight must eliminate all potential testing artifacts, including inadequate fixation, light scatter, nonvisual cues, and nonrandom presentation of targets.
Even if blindsight does exist, it is clear that visual discriminations in blind hemifields are less accurate and more variable than those in normal hemifields. However, the range of visual function demonstrated in patients with either blindsight or residual vision is impressive, including perception of spatial location and discrimination of form, orientation, color, and motion. No clear pattern of visual abilities that are preserved or destroyed has emerged in such patients. The variability in blindsight profiles may have an anatomic basis in the inevitable variability of naturally occurring human lesions. Most lesions of striate cortex also involve some extrastriate regions, but the degree and the areas affected differ from one patient to the next. The fact is, not all patients with cortical field defects have blindsight. Why some patients have blindsight and others do not is unclear. Its presence or absence may reflect differences in lesion anatomy, but this is not proven. Some studies have found that blindsight was present only in patients with hemianopia occurring in childhood, a period of presumably greater neural plasticity, but other studies have not confirmed this impression. A requirement for childhood onset is inconsistent with reports that training in older patients can lead to blindsight. If blindsight truly exists, it is uncommon.
Cerebral Achromatopsia
Cerebral achromatopsia, also called central achromatopsia, is an uncommon defect of color perception caused by damage to the visual cortex. Some patients with this condition complain that colors look dull, wrong, or less bright, whereas others report that their world is completely colorless and that objects appear only in shades of gray, like in old black and white movies. Although the term “cerebral achromatopsia” is sometimes used to include all degrees of cerebral color deficits, we believe that this term is best reserved for the most severe cases and that the term cerebral dyschromatopsia should be used in cases where there is residual color sensation.
Either cerebral achromatopsia or cerebral dyschromatopsia may be a presenting symptom or may evolve during recovery from cortical blindness. The most common setting is vertebrobasilar ischemia affecting the posterior cerebral arterial blood supply to the occipital lobes. Other causes of achromatopsia include herpes simplex encephalitis, cerebral metastases, recurrent focal seizures, and dementia with visual cortical involvement. Transient achromatopsia may also occur as part of the aura of migraine.
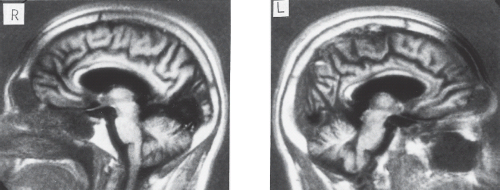 Figure 13.2 Magnetic resonance imaging in a patient with cerebral dyschromatopsia. Nonenhanced sagittal T1-weighted images show bilateral lesions in the visual cortices. The lesion of the right hemisphere (R) is responsible for the left homonymous hemianopia. The lesion of the left hemisphere (L) is located below the calcarine fissure in the inferior visual association cortex in regions thought to process color. (Reprinted with permission from Rizzo M, Nawrot M, Blake R, et al. A human visual disorder resembling area V4 dysfunction in the monkey. Neurology. 1992;42:1175–1180.) |
Anatomic and functional imaging studies indicate that color is processed by a large network of structures, including V1, V2, V3, V4 (the most emphasized), and IT. Nevertheless, the lesions causing cerebral achromatopsia are rather restricted and usually affect the ventromedial sectors of the occipital lobe in the lingual and fusiform gyri (Fig. 13.2). Lesions of the middle third of the lingual gyrus or the white matter immediately behind the posterior tip of the lateral ventricle
are said to be critical. Bilateral lesions are necessary for complete achromatopsia; unilateral right or left occipital lesions may produce a hemiachromatopsia.
are said to be critical. Bilateral lesions are necessary for complete achromatopsia; unilateral right or left occipital lesions may produce a hemiachromatopsia.
Common accompaniments of cerebral achromatopsia include superior homonymous quadrantanopia, visual agnosia, and acquired alexia. The quadrantanopia may be complete or incomplete; but in either case, the achromatopsia affects the remaining inferior quadrants of the visual fields on the side opposite the lesion (i.e., on the same side as the visual field defect). Visual agnosia—the inability to recognize previously familiar objects or to learn the identity of new objects by sight alone despite adequate visual sensory abilities—is also seen in patients with cerebral achromatopsia, particularly in patients with bilateral lesions. The syndrome in such patients includes prosopagnosia in which the agnosic defect is most striking for faces; and topographagnosia or topographic disorientation in which patients tend to get lost in familiar visual surroundings, partly because of an inability to recognize previously familiar local landmarks. Some patients have pure alexia, also known as alexia without agraphia, the acquired inability to read in previously literate individuals (see below). Defects of visual memory and even generalized amnesia can accompany cerebral achromatopsia, depending on the extent of the lesion into more anterior and mesial structures in the temporal lobes.
One should be cautious in interpreting the verbal report of a patient with cerebral achromatopsia. Assessment of color naming and associations should be conducted independently of color perception testing. This allows the best opportunity to distinguish cerebral achromatopsia and dyschromatopsia from color anomia, agnosia, and related defects of language and memory. Using matching tasks in addition to naming tasks is crucial.
Color anomia is often part of a more general anomia in aphasic patients, but there are instances in which color naming is disproportionately affected. These cases tend to occur with left occipital lesions and are associated with a complete right homonymous hemianopia, rather than just the upper quadrantanopia seen with cerebral achromatopsia. One possible mechanism in such cases is an interhemispheric disconnection syndrome associated with right homonymous hemianopia and pure alexia. Because of the complete right homonymous hemianopia, visual input to language areas in the left hemisphere must arise from the right visual cortex. However, a concurrent lesion of the splenium of the corpus callosum interrupts this process. Affected patients can perceive colors yet not name them (color anomia) and may also see letters but not be able to read them (pure alexia). Preserved tactile naming in such patients suggests preservation of callosal fibers anterior to those required for reading and color naming. Color anomia, together with pure alexia, can also occur without a callosal lesion when there is damage near the posterior aspect of the occipital horn on the left, interrupting connections from both visual fields to language areas.
Prosopagnosia and Related Disturbances of Object Recognition
Visual agnosia is an inability to recognize familiar objects, despite adequate visual perception attention, intellect, and language. Prosopagnosia is a restricted form of visual agnosia characterized by the impaired ability to recognize familiar faces or to learn to recognize new faces. It is a specific functional disorder with a specific neuroanatomic basis. Prosopagnosia may actually cover a spectrum of deficits, with individual patients varying in the degree of perceptual versus memory dysfunction or even having disconnections between the two processes. Furthermore, abnormal face recognition may be part of more generalized perceptual, cognitive, or memory problems. We will reserve the term “prosopagnosia” for cases in which the facial recognition deficit is disproportionately more severe than other associated deficits.
Patients with prosopagnosia are usually aware of their difficulty and complain of the social embarrassment of not recognizing acquaintances. When they do recognize others, it is often by reliance on specific facial features or paraphernalia (e.g., a hairstyle, glasses, beards and mustaches, a missing tooth, or a particular scar) that bypass the need for an overall analysis of the shape of the face. They may also use nonfacial visual cues, such as gait and posture, or nonvisual cues, such as voice. The context of the encounter is also important. For example, patients with prosopagnosia may be able to recognize hospital staff but are unable to recognize the same persons when they meet them later on the street. Persons with childhood onset of prosopagnosia and, occasionally, persons with onset in adulthood may be ignorant of their deficit.
Despite abnormal recognition of familiar faces, many patients with prosopagnosia can accurately discriminate among unfamiliar faces, even with varying lighting conditions or facial views (i.e., Benton Facial Recognition Test [BVRT]). However, patients with prosopagnosia may require much more time than normal to arrive at correct judgments, suggesting that they use an inefficient and abnormal route for processing face information.
Some individuals with prosopagnosia can judge the age, gender, and emotional expression of faces they cannot recognize, and some even lip-read. Again, prosopagnosics may use different perceptual processes from those used by normal individuals to make such
judgments. For example, a patient with prosopagnosia might rely on wrinkles to tell age, whereas normal individuals can still make age judgments in the absence of such local features. Other prosopagnosic patients, usually those with more pervasive perceptual dysfunction, are impaired with respect to judgments about facial age, sex, emotional expression, and the direction of the person’s gaze, the last item being an important social signal.
judgments. For example, a patient with prosopagnosia might rely on wrinkles to tell age, whereas normal individuals can still make age judgments in the absence of such local features. Other prosopagnosic patients, usually those with more pervasive perceptual dysfunction, are impaired with respect to judgments about facial age, sex, emotional expression, and the direction of the person’s gaze, the last item being an important social signal.
An important theoretical issue is whether the prosopagnosic defect is specific for faces or also affects perception of other objects. Some patients have difficulty distinguishing types of objects, such as makes of cars, flowers, food, and coins. They also have trouble identifying previously familiar items, such as buildings, handwriting, and personal belongings or clothing. These cases imply that in prosopagnosia, impaired facial recognition may simply be the most prominent manifestation of a more general recognition or perceptual problem. On the other hand, some patients can distinguish nonface objects relatively well despite severe prosopagnosia. Specific tests of face versus object recognition appear to confirm this dissociation.
Although patients with prosopagnosia deny familiarity with and cannot identify known faces, some patients retain covert (i.e., nonconscious) recognition of these faces at a variety of levels. Investigators have shown two main phenomena: Covert familiarity, in which the patient is able to distinguish previously familiar but unrecognized faces from completely unknown faces, and covert semantic knowledge, in which the patient retains information about name, occupation, and other facts associated with a face.
Patients with prosopagnosia usually have difficulty recognizing faces of people with whom they were familiar before the start of the illness (retrograde prosopagnosia) as well as people whom they meet after the onset of the condition (anterograde prosopagnosia). Some patients have slightly better recognition of people long known to them, whereas others have only anterograde prosopagnosia. Prosopagnosia historically has been segregated into two main forms: Apperceptive prosopagnosia, in which there is a failure to form a sufficiently accurate facial percept; and associative prosopagnosia, in which there is an inability to match an accurate percept to facial memories.
In most cases of prosopagnosia, the lesion is in the inferior temporo-occipital cortex, usually in the lingual and fusiform gyri (Fig. 13.3). In occasional cases, the lesion appears to be located in the more anterior temporal cortex. All autopsied cases of prosopagnosia have bilateral lesions, often symmetric, presumably affecting homologous regions of both hemispheres. However, the position of the left-sided lesion can be more variable. A left-sided hemispheric lesion or a lesion of the splenium of the corpus callosum might disconnect a right hemispheric locus for facial recognition from visual input of the right hemifield, with critical effects in cases with left homonymous hemianopia. Furthermore, reports of neuroimaging showing apparently unilateral right temporo-occipital lesions have accumulated. The type of face perception deficits caused by unilateral as opposed to bilateral cerebral lesions may differ. It has been suggested that apperceptive types of prosopagnosia occur with damage to right ventral and dorsal temporo-occipital areas, whereas bilateral ventral temporo-occipital lesions result in an associative or dysmnesic form.
Almost all patients with prosopagnosia have other visual deficits. Visual field defects are frequent, the most common being left homonymous hemianopia, left superior homonymous quadrantanopia, bilateral superior quadrantanopia, or combinations of these. The visual field defect, achromatopsia or hemiachromatopsia, and topographagnosia often form a common tetrad with prosopagnosia. Nevertheless, cases of prosopagnosia associated with normal color vision exist, showing that prosopagnosia and achromatopsia are dissociable. Visual object agnosia is said to be absent in some patients with prosopagnosia and present, though proportionately milder, in others.
A frequent finding in patients with prosopagnosia is impaired visual memory. In some of these patients, the memory deficit also affects verbal material. Other patients with prosopagnosia also have simultanagnosia, palinopsia, visual hallucinations, constructional difficulties, and left hemineglect. Hemisensory deficits or hemiparesis occur in some patients with unilateral right-sided lesions, although in some of these cases, the motor and sensory findings are related to other lesions. Pure alexia (see below) can occur in some patients with prosopagnosia.
The most common lesions causing prosopagnosia are posterior cerebral artery infarctions and, less often, viral encephalitis. The predominance of these conditions may be related to their potential to inflict bilateral damage, although the neuroimaging in some cases shows only a unilateral lesion. Other unilateral lesions, such as tumors, hematomas, abscesses, and surgical resections, are less frequently reported.
Impaired facial recognition can occur as part of the generalized dementia in Alzheimer disease, in Parkinson disease, and rarely in elderly patients with bilateral or unilateral right temporal lobar degeneration. The last disorder is one of the focal progressive atrophies, others of which affect the frontal, parietal, or left temporal lobes possibly related to Alzheimer or Pick disease.
Developmental prosopagnosia also has been described. Patients with this condition usually are not aware that they have problems with face recognition until they experience social difficulties related to the impairment. In addition to problems with
facial recognition, these patients have difficulty judging facial age, sex, and expressions. They also perform facial matching tasks very slowly, suggesting a perceptual process differing from normal. In some cases, developmental prosopagnosia is present in other family members, possibly as an autosomal-dominant trait, and coexists with the Asperger syndrome of autism. The prevalence of prosopagnosia among all autistic individuals is unknown, as are the effects of the face perception disorder on other aspects of psychosocial development.
facial recognition, these patients have difficulty judging facial age, sex, and expressions. They also perform facial matching tasks very slowly, suggesting a perceptual process differing from normal. In some cases, developmental prosopagnosia is present in other family members, possibly as an autosomal-dominant trait, and coexists with the Asperger syndrome of autism. The prevalence of prosopagnosia among all autistic individuals is unknown, as are the effects of the face perception disorder on other aspects of psychosocial development.
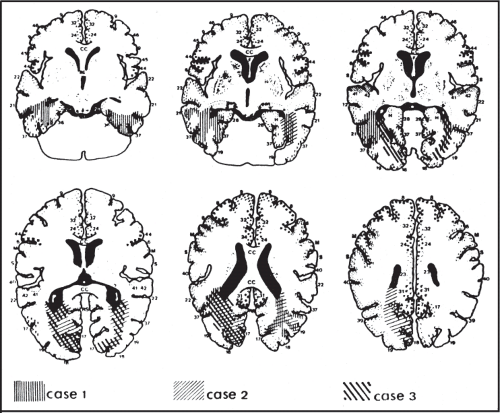 Figure 13.3 Location of bilateral lesions in three patients with prosopagnosia. Axial template drawings with shaded areas representing the lesions of three patients with prosopagnosia. Bilateral damage, greater on the left than the right, is present in all three cases. (Reprinted with permission from Damasio AR, Damasio H, van Hoesen GW. Prosopagnosia: Anatomic basis and behavioral mechanisms. Neurology. 1982;32:331–341.) |
Acquired Alexia
Acquired alexia is the loss of efficient reading for comprehension, despite adequate visual acuity. Reading is a complex behavior involving perception of form, spatial attention, ocular fixation, scanning saccadic eye movements, and linguistic processing. Not surprisingly, many types of cerebral or visual dysfunction can disrupt the reading process. Although other clinical signs may overshadow the difficulty, impaired reading is sometimes the chief complaint. The severity of reading difficulty can range from a mild defect with slow reading and occasional errors, which may only be identified by comparison with normal controls matched for educational level, to a complete inability to read even numbers and letters. Thus, “dyslexia” is the more appropriate term in most cases, and it is used in this manner (and not to describe a learning disorder) in this chapter. Analysis of not only the severity but also the type of reading errors can help differentiate among the various forms of reading impairment and their causes.
Most syndromes of alexia can be explained by the disconnectionist theories. The left
angular gyrus stores the visual representation of words necessary for reading and writing. Therefore, disconnecting the visual inputs of both hemispheres from the left angular gyrus could disrupt reading but leave writing intact—that is, pure alexia.
angular gyrus stores the visual representation of words necessary for reading and writing. Therefore, disconnecting the visual inputs of both hemispheres from the left angular gyrus could disrupt reading but leave writing intact—that is, pure alexia.
The key feature of pure alexia (alexia without agraphia, or word blindness), is a dramatic dissociation between the ability to read and the ability to write. Patients with this condition can write fluently and spontaneously, but having done so are unable to read what they have just written. The severity varies from a slow, laborious reading of words one letter at a time (letter-by-letter reading; spelling alexia) to complete inability to read words or letters (global alexia) and sometimes even numbers or other symbols. Covert reading ability can be detected in at least some patients with pure alexia. These patients have aspects of preserved reading ability of which they are not aware.
Lesions causing pure alexia are almost always located in the left hemisphere, most commonly in the inferior temporo-occipital region (Fig. 13.4




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
