Purpose
To provide a detailed review of current clinical guidelines for the diagnosis, work-up and treatment of autoimmune retinopathy and to preview briefly possible future therapies.
Design
Perspective based on literature review and clinical expertise.
Methods
Interpretation of current literature, relying on the authors’ clinical experience.
Results
Autoimmune retinopathy is a rare immunologic disease characterized by the presence of circulating antiretinal antibodies along with electroretinographic and visual field abnormalities. An ophthalmic examination can be normal or show minimal findings. The diagnosis of autoimmune retinopathy is made difficult by diagnostic criteria that are both limited and nonstandardized. Currently, the diagnosis is made based on the demonstration of serum antiretinal antibodies and the presence of clinical manifestations (including abnormal electroretinographic findings). The mere presence of these antibodies is not diagnostic. Lack of an accepted gold standard for antiretinal antibodies detection and poor interlaboratory concordance make the diagnosis challenging. There are anecdotal reports of immunosuppressive therapy in autoimmune retinopathy; however, the response to treatment is variable, with more favorable results achieved in paraneoplastic retinopathy, particularly cancer-associated retinopathy, with a combination of chemotherapy and immunosuppression. Whether an earlier attempt to treat nonparaneoplastic autoimmune retinopathy would be more beneficial is unknown. Early treatment attempts are limited by lack of sensitive and specific assays and definitive clinical criteria.
Conclusions
Little is known about the clinical course, prognosis and treatment of autoimmune retinopathy. Additional studies should examine the specificity and pathogenicity of antiretinal antibodies and screen for biomarkers, and they should be conducted concurrently with studies seeking to identify appropriate treatment.
Autoimmune retinopathy is an inflammation-mediated retinopathy characterized by vision loss, scotomas, visual field deficits, photoreceptor dysfunction, and the presence of circulating antiretinal antibodies. On clinical examination, the fundus usually appears unremarkable; however, some patients may show retinal pigment epithelium abnormalities, vascular attenuation or optic disc pallor. There is minimal or no intraocular inflammation. The sine qua non of autoimmune retinopathy is the presence of circulating antiretinal antibodies, which target retinal antigens and are believed to be responsible for the photoreceptor damage, though the precise mechanisms are not entirely understood. Autoimmune retinopathy can be divided into 2 groups: paraneoplastic and nonparaneoplastic, with paraneoplastic further subdivided into cancer-associated retinopathy (CAR) and melanoma-associated retinopathy (MAR). Nonparaneoplastic autoimmune retinopathy is probably more common than paraneoplastic retinopathies. CAR is more common than MAR, though the prevalence of MAR is increasing, while CAR prevalence is decreasing. Vision loss and photoreceptor dysfunction associated with cancer was first described by Sawyer et al in 1976 and the term paraneoplastic retinopathy was coined by Klingele et al. in 1984.
Although it is believed to be rare, the prevalence of autoimmune retinopathy is currently unknown. It constitutes far less than 1% of all cases seen at our tertiary uveitis and ocular immunology clinic. The overlap of clinical features with other degenerative retinal disorders and lack of standardized clinical and laboratory diagnostic criteria may be contributing to an underestimation of its prevalence. In this article we focus on pathophysiology, clinical manifestations and management of the nonparaneoplastic form of autoimmune retinopathy.
Pathophysiology
Multiple retinal proteins have been found to be antigenic; some of these are retina-specific (eg, recoverin), and others can be found in nonretinal tissues as well (eg, α-enolase). Although recoverin, a 23kDa calcium-binding protein found in photoreceptors, and α-enolase, a 48kDa ubiquitous glycolytic enzyme, are the most widely studied antigens in autoimmune retinopathy, associations with autoantibodies against carbonic anhydrase, arrestin, transducin-β, TULP1, neurofilament protein, heat shock protein-70, photoreceptor-cell–specific nuclear receptor, Müller-cell-specific antigen, transient receptor potential cation channel, subfamily M, member 1 (TRPM1), and a number of yet unidentified putative antigen targets have been reported . Evidence suggests that paraneoplastic autoimmune retinopathy may be triggered by molecular mimicry between tumor antigens and retinal proteins. Using immunohistochemical staining, serum from patients with CAR labeled photoreceptors in human retinal sections and reacted with a 23 kDa protein on Western blot (WB). The antigen was later identified as recoverin, which is a calcium-binding protein found in photoreceptors and has been shown to be expressed in the tumor cells of patients with cancer-associated retinopathy. A similar mechanism has been suggested in anti-alpha-enolase–mediated CAR. It is possible that nonparaneoplastic forms may also be triggered by a cross-reaction between retinal proteins and presumed viral or bacterial proteins. Recoverin is most commonly associated with CAR but has also been found in nonparaneoplastic autoimmune retinopathy as well. Similarly, α-enolase has been associated with both paraneoplastic and nonparaneoplastic forms.
Both in vitro and in vivo experiments have attempted to elucidate the pathogenic role of antiretinal antibodies. In vitro studies have shown that both recoverin and α-enolase induce apoptosis of retinal cells following cellular internalization via caspase pathways and intracellular calcium influx. An in vivo experiment in monkey eyes showed that intravitreal injection of human MAR IgG altered the b-wave in monkey electroretinographic (ERG) findings, mimicking the ON-bipolar cell dysfunction and the negative ERG findings commonly seen in patients with MAR. This experiment supports the hypothesis that circulating MAR IgG plays a role in MAR pathogenicity. In spite of this evidence supporting the pathogenic role of antiretinal antibodies, it is still unclear why some patients with such antibodies develop retinopathy while others do not.
Antiretinal antibodies can target any retinal cell-type, including photoreceptor cells, ganglion cells and bipolar cells. However, the presence of these antibodies alone is not sufficient for the diagnosis of this ocular disorder because they can also be found in a variety of retinal diseases and systemic autoimmune diseases as well as in the serum of healthy individuals.
Clinical Features and Diagnosis
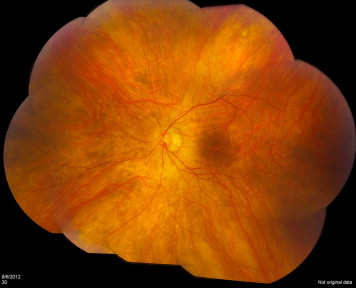
Patients with nonparaneoplastic autoimmune retinopathy typically present with subacute vision loss, scotomas, photopsias, nyctalopia, or photoaversion, and dyschromatopsia. Visual acuity can be deceivingly good in the early stages. On examination, the fundus may appear unremarkable or demonstrate retinal vascular attenuation, diffuse retinal atrophy, changes in the RPE, and waxy disc pallor ( Figure 1 ). The disease is usually bilateral but it can be asymmetric. Typically there are minimal or no intraocular inflammatory cells. Among nonparaneoplastic patients, there is a predominance of females (63% to 66%), and histories of autoimmune disease are common. The typical patient with autoimmune retinopathy would be an adult female in her fifth to sixth decade with no history of visual problems prior to the onset of photopsias, the presence of scotomas, and no family history of retinitis pigmentosa (RP). If these features, and the circulating antiretinal antibodies are present, and if there is no malignancy at presentation or following a thorough investigation, a tentative diagnosis can be made.
In an effort to simplify and standardize the diagnostic criteria for nonparaneoplastic autoimmune retinopathy, the authors propose a set of 4 essential criteria along with 5 symptoms that serve as supportive criteria. Essential elements include no evidence of malignancy after a thorough work-up; no evidence of degenerative eye disease such as RP; a positive screen for serum antiretinal antibodies; and an abnormality in ERG findings with or without visual field abnormality. Supportive criteria include the presence of symptoms, such as photopsias, scotomas, nyctalopia or photoaversion, and dyschromatopsia.
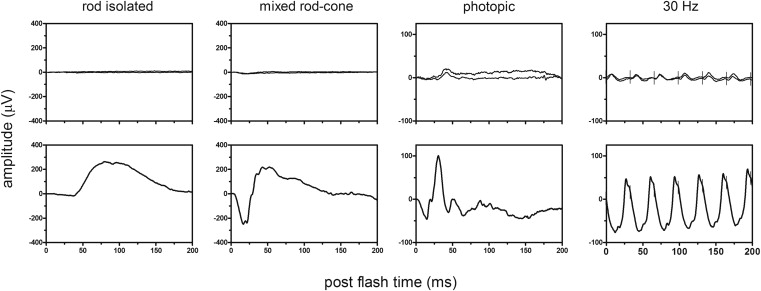
Visual-field testing shows constriction and central or paracentral scotomas, and ERG can show abnormalities in dark-adapted or light-adapted responses, bipolar cell responses or a combination of these responses. Fluorescein angiography in autoimmune retinopathy rarely shows leakage in the macula, and optical coherence tomography (OCT) can show cystoid macular edema, typically in the form of cystic spaces. Recent advances in imaging technology are promising. For instance, OCT and fundus autofluorescence (FAF) in patients with autoimmune retinopathy showed abnormal autofluorescence patterns, mainly in the form of an hyperautofluorescent ring in the parafoveal region that corresponds to loss of outer-retinal structures on spectral domain OCT (SD-OCT). Loss or disruption of the photoreceptor layer with decreased central macular thickness have also been observed. Both FAF and OCT have the potential to aid in the diagnosis, to understand its pathogenesis, and to monitor disease progression ( Figures 2-5 ). In our experience with 24 patients who had nonparaneoplastic autoimmune retinopathy, the most common findings were loss of the inner/outer segment layer on SD-OCT and parafoveal hyperautofluorescent ring or at least mild speckling on FAF, which were present in approximately half of our patients (unpublished data).
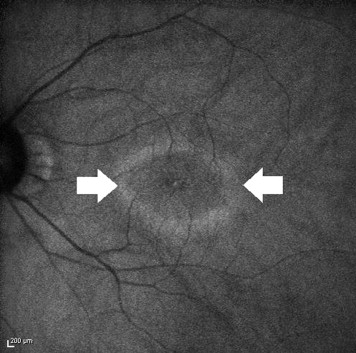
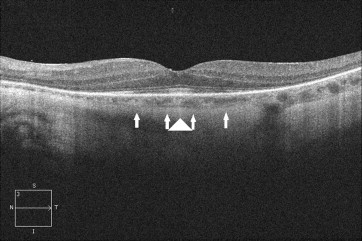
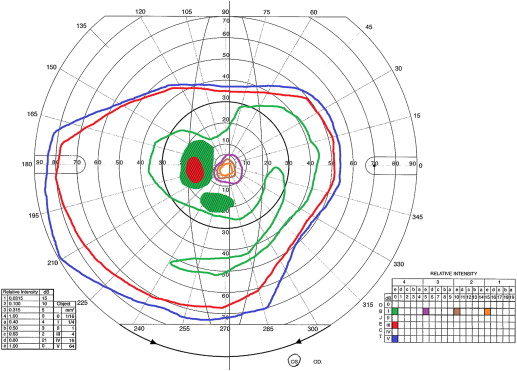
As might be expected for an entity with no consensus in diagnosis, retrospective studies in patients with nonparaneoplastic autoimmune retinopathy showed that clinical features vary considerably. In one study, diffuse retinal atrophy was seen in the majority of patients (83%) and pigment deposits in only a small proportion (13%); macular edema was present in approximately half of these cases. In another study, pigmentary changes were seen in approximately half of the patients, and macular edema was present in only 24%. Given the nature of our referral center, the majority of our patients had more advanced disease and demonstrated clinical findings such as RPE mottling, pigment deposits, and attenuated vessels (unpublished data).
Demonstration of antiretinal antibodies is crucial for the diagnosis of autoimmune retinopathy. They can be detected using WB, immunohistochemistry (IHC) or enzyme-linked immunosorbent assay (ELISA). Each approach has its corresponding advantages and disadvantages, and the most commonly performed techniques tend to be WB and IHC. WB identifies antibodies based on the size of the protein and is both technically difficult and lacking in specificity. For example, the detection of a 23 kDa band on WB does not necessarily mean antibody against recoverin. Using IHC to detect antiretinal antibodies, on the other hand, involves fixing patient serum against frozen retina from a human donor or monkey or mouse. Sections are then analyzed using light microscopy to determine which layers of the retina the antibody binds to. The advantage of IHC is the ability to localize the specific site of binding within the retina. ELISA involves adding various dilutions of patient sera into wells coated with specific retinal antigens, and binding is detected using secondary antibodies. The clear disadvantage of this approach is that one must know the antigen of interest for a specific antibody ahead of time, thus it lacks sensitivity in identifying all potential antiretinal antibodies. Unfortunately, all of these techniques lack standardization. Furthermore, it must be emphasized that the mere detection of antiretinal antibodies is not sufficient for a diagnosis of autoimmune retinopathy, nor does it prove that the antibodies detected are pathogenic. Currently, the only commercially available testing is through Oregon Health Sciences University (OHSU) (Available at http://www.ohsu.edu/xd/health/services/casey-eye/clinical-services/diagnostic-services/upload/Ocular-Immunology-Web-2.pdf ).
A recent study examined the concordance rate of antiretinal antibody testing between 2 separate laboratories that commonly accept samples from outside their own institutions. They point out that though the sample shipment and handling were performed identically for both laboratories, the processing and detection methods employed by the laboratories are distinct. Laboratory A used human retinal extract with positive controls (serum samples of patients with a known antiretinal antibody) and negative controls (using only secondary antibody). Laboratory B used pig retinal extract with normal controls (serum samples of people with no antibody activity). Of note, they do not mention whether the dilutions used were the same at both laboratories, an aspect that would undoubtedly affect the sensitivity of antibody detection. This study found that the overall concordance rate of any antiretinal antibody detection was 60% in the 2 laboratories, and that among that rate, just over half, or a mere 36% of the total cohort, showed antiretinal antibody-specific concordance (same band detected on WB). The overall interobserver agreement was very poor, with a kappa value of −0.13. We agree with the authors’ conclusion that the lack of a gold standard has led to dramatic variability in laboratory detection of antiretinal antibodies, and standardized methods across laboratories are urgently needed in order to produce consistent results.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


