Anterior Corneal Dystrophies: Dystrophies of the Epithelium, Epithelial Basement Membrane, and Bowman’s Layer
Anterior Corneal Dystrophies: Dystrophies of the Epithelium, Epithelial Basement Membrane, and Bowman’s Layer
Puwat Charukamnoetkanok
Suhas Tuli
Dimitri T. Azar
Corneal dystrophies are noninflammatory, hereditary disorders that principally involve the central cornea and appear to have little or no relationship to systemic or environmental factors. They are classified according to the anteroposterior localization of the major pathologic alteration in the cornea: epithelial, stromal, and posterior dystrophies (
Table 45-1) (
1). This chapter focuses on the anterior dystrophies affecting the epithelium, epithelial basement membrane, and Bowman’s layer. The study of the pathogenesis of corneal dystrophies at the molecular level has increased our understanding of the genetic basis of corneal dystrophies. A gene-based reclassification of corneal dystrophies may be possible in the near future (
2). At present, however, the current classification scheme, based on the clinical and histologic findings, remains useful for the management of patients suffering from these potentially blinding corneal disorders.
JUVENILE HEREDITARY EPITHELIAL DYSTROPHY (MEESMANN’S DYSTROPHY)
Meesmann’s dystrophy is a bilateral, diffuse, symmetric corneal dystrophy involving accumulation of intracytoplasmic deposits in the corneal epithelium and manifesting as uniformly sized epithelial cysts.
The disorder was first described clinically by Pameijer (
3) in 1935 and histopathologically by Meesmann and Wilke (
4) in 1939. This rare autosomal-dominant dystrophy (
5) has incomplete penetrance (as low as 60%) and variable expressivity (
3,
4,
5,
6,
7,
8,
9). However, Stocker and Holt (
5) also reported a recessive form in 20 individuals in a group of 200 descendants of Moravian settlers. The molecular basis of Meesmann’s epithelial corneal dystrophy has been attributed to mutations in two causative genes, the cornea specific keratin genes
KRT3 (chromosome 12q13) and
KRT12 (chromosome 17q12), which encode cytoskeletal proteins K3 and K12, respectively (
10,
11,
12). The mechanisms by which these mutations cause the Meesmann’s phenotype remain to be elucidated.
Clinical Features
Corneal involvement is bilateral and is most prominent in the interpalpebral zone. Clusters of tiny round cysts in the epithelium appear as discrete white spots on focal illumination and as refractile, clear, cystlike structures on retroillumination (
Fig. 45-1) (
8). These myriad fine round cysts of uniform size and shape usually become visible by 12 months of age and increase in number throughout life. Coalescence of cysts may form refractile lines. Patients remain asymptomatic until the fourth or fifth decades of life, when photophobia, redness, and pain occur because of recurrent erosions due to rupture of cysts. Subepithelial gray opacities appear at the late stage of the disease. In general, visual acuity remains functionally good, but patients may complain of blurred vision (
5).
Histopathology
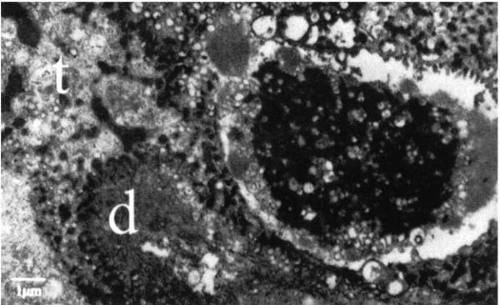
The primary pathosis in Meesmann’s dystrophy is in the basal epithelium, with accumulation of an abnormal, electron-dense, intracytoplasmic material described as a “peculiar substance” (
Fig. 45-2). This fibrillogranular material, whose exact composition is unknown, causes cell death. Cysts are actually areas of cellular degeneration and contain periodic acid-Schiff (PAS)-positive material (
Fig. 45-3). Increased cell turnover results in thickening of the basement membrane and increased glycogen content of the basal epithelial cells (
6,
13,
14). Bowman’s layer and the superficial stroma are unaffected.
Management
Because the patients are usually asymptomatic or have minimal symptoms, treatment is rarely required. Debridement of the epithelium is usually followed by reappearance of symptoms. Although curative, superficial keratectomy is rarely warranted (
15). Excimer laser phototherapeutic keratectomy (PTK) has emerged as an alternative treatment of recalcitrant recurrent erosions. With PTK the epithelial defect sites are ablated to a depth of 3 to 4
μm with reportedly promising results (
16,
17,
18,
19,
20,
21,
22,
23,
24). Lamellar and penetrating keratoplasty are seldom indicated (
5,
13). Although the dystrophy can recur in grafts, the recurrent disease is often less severe.
EPITHELIAL BASEMENT MEMBRANE DYSTROPHY (COGAN’S MICROCYSTIC DYSTROPHY)
Epithelial basement membrane dystrophy (EBMD) is a corneal disorder characterized by recurrent epithelial erosion as a result of abnormal epithelial turnover, maturation, and production of basement membrane. The lesions are bilateral but can be asymmetric. The findings are variable and may change with time.
This dystrophy is also known by many other names: fingerprint/map/dot dystrophy, fingerprint dystrophy, anterior basement dystrophy, and dystrophic recurrent erosion. The
underlying pathology is localized to the abnormal epithelial-basement membrane adhesion complex and manifests clinically as recurrent erosions. The primary alteration in the epithelial-basement membrane adhesion complex could be due to a defect of the epithelium, the epithelial basement membrane, or Bowman’s membrane, in combination with or without production of abnormal substances.
Clinical Features
EBMD is the most frequently encountered anterior corneal dystrophy in clinical practice (
15,
25,
26,
27,
28). Although autosomal-dominant forms have been reported (
29,
30,
31), many cases do not have a definite hereditary pattern. Changes similar to fingerprint/map/dot dystrophy are seen in about 6% of the normal population above the age of 50 years (
32).
Cogan et al. (
25) first described microcystic dystrophy of the cornea in five unrelated women at the Massachusetts Eye and Ear Infirmary in Boston, who had bilateral grayish-white spherical lesions of varying sizes. Maplike dystrophy was described by Guerry in 1965 (
33), who had earlier reported fingerprint lines (
34).
The patients with EBMD are predominantly white women above 30 years of age. But EBMD can become manifest as early as 4 to 8 years of age in familial cases. In nonfamilial cases, recurrent erosions occur for a few years, improve spontaneously, and do not leave significant residual visual acuity loss (
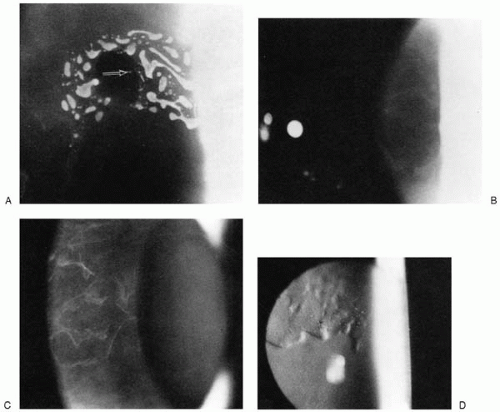
35). Maps are geographic circumscribed gray lesions best seen with broad oblique illumination. They appear in varying shapes and sizes and do not stain with fluorescein (
Fig. 45-4). Dots are cysts or fine blebs seen as gray-white intraepithelial opacities of various sizes in close proximity to the maplike patches (
Fig. 45-5). Fingerprint lines are branching refractile lines with club-shaped terminations (
Fig. 45-6) (
31). Retroillumination will strikingly highlight these lesions. Leashes of long, thick fingerprint lines are called “mares tails.” Combinations of map and dots are encountered most frequently, followed by maps alone. Dots are the least frequently encountered of the three lesions. It is rare that fingerprints are present along with maps and dots, whereas dots alone are never seen. Visual acuity is usually minimally affected. However, irregular astigmatism or higher-order aberration may lead to visual loss.
Histopathology and Pathogenesis
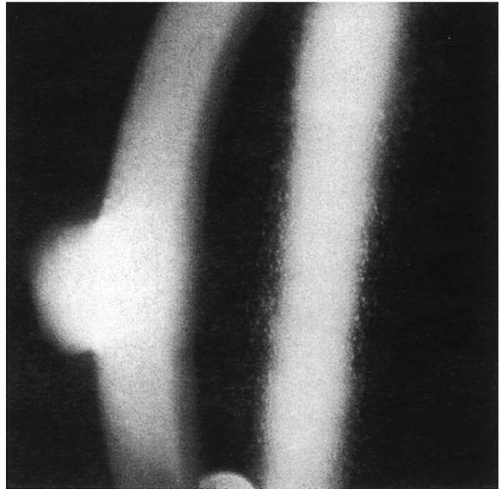
The basic pathology in EBMD is the synthesis of abnormal basement membrane that is responsible for the whole spectrum of clinical manifestations. Erosions are due to absence
of hemidesmosomal connections of the epithelial cells in areas with abnormal basement membrane. Maps are projections of abnormal multilaminar basement membrane into the epithelium (
Fig. 45-7) (
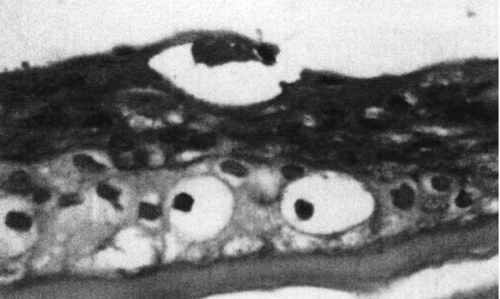
36). Dots are pseudocysts filled with lipid and cytoplasmic and nuclear debris (
Fig. 45-8). Cysts form in areas with intraepithelial extensions
of aberrant basement membrane. Epithelial cells beneath the aberrant basement membrane become vacuolated and liquefied. Rupture of these cysts causes recurrent erosions. Fingerprints represent linear projections of fibrillogranular material into the epithelium with thickening of the basement membrane (
Fig. 45-9).


 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access
