, Yasuya Nomura2 and Yasuya Nomura3
(1)
The Society for Promotion of International Oto-Rhino-Laryngology, Tokyo, Japan
(2)
The University of Tokyo, Tokyo, Japan
(3)
Showa University, Tokyo, Japan
Abstract
This chapter discusses cochlear hyperplasia, hypophosphatasia, Alagille syndrome, Scheibe dysplasia, and Mondini dysplasia. Anomalies of the inner ear usually derive from arrested embryonic organ development. Cochlear hyperplasia results from excessive development of the organ of Corti and cochlear duct. Because the bony cochlea is normal in shape, the overdeveloped cochlear duct with the organ of Corti reverses at its apical end and heads backwards into the scala tympani, producing a mirror image of the organ of Corti. The case of hypophosphatasia described here showed immature ossification of the bony labyrinth with abnormal tissue formation in the lateral wall of the membranous cochlea. The parents of this patient were consanguineous and their sera showed low alkaline phosphatase activity. Alagille syndrome is a systemic malformation, in which the posterior semicircular canal shows a partial deficit. In cases of Scheibe dysplasia the bony labyrinth develops normally, but the cochleosaccular portion of the membranous labyrinth is dysplastic. Mondini dysplasia is relatively common, but no two cases are identical in detail.
Keywords
Alagille syndromeCochlear hyperplasiaHypophosphatasiaMondini dysplasiaScheibe dysplasia9.1 Cochlear Hyperplasia
9.1.1 Introduction
Various anomalies may be seen in the cochlea of humans and other animals. Jackler et al. [1] classified congenital malformations of the cochlea as follows:
1.
Complete labyrinthine aplasia (Michel deformity)
2.
Cochlear aplasia: no cochlea, normal or malformed vestibule and semicircular canals
3.
Cochlear hypoplasia: small cochlear bud, normal or malformed vestibule and semicircular canals
4.
Incomplete partition: small cochlea with incomplete or no interscalar septum, normal or malformed semicircular canals
5.
Common cavity: cochlea and vestibule form a common cavity without internal architecture; normal or malformed semicircular canals
These malformations result from arrested maturation during one of the stages of inner ear embryogenesis. Other malformations may result from aberrant development.
9.1.2 Cochlear Hyperplasia
In a series of experiments using guinea pig cytomegalovirus (GPCMV), Nomura et al. [2] found a strange cochlear anomaly: hyperplasia of the organ of Corti with the scala media. They inoculated GPCMV into the cochlea of a female guinea pig through the round window membrane. Shortly after inoculation the animal became pregnant and 2 months later bore a litter of four. One of the offspring showed an anomalous right cochlea. The bony cochlea had normal coils. The organ of Corti, however, did not end at the apex, but together with the scala media continued backwards into the scala tympani. Graphic reconstruction revealed that the cochlea had a total length of 24 mm, including an overgrowth of 1 mm (Fig. 9.1). Therefore, the organ of Corti showed a symmetrical mirror image, sharing a common scala tympani at the apical turn (Fig. 9.2). The inverted organ of Corti and scala media intruded into the space normally occupied by the scala tympani. The outer hair cells seemed excessive in number and the Deiters’ cells beneath these hair cells were either hypoplastic or absent (Figs. 9.3 and 9.4). The limbus spiralis, inner sulcus, osseous spiral lamina, and nerve fibers were hypoplastic in the inverted portion. The inner ear structures other than the cochlea were normal.
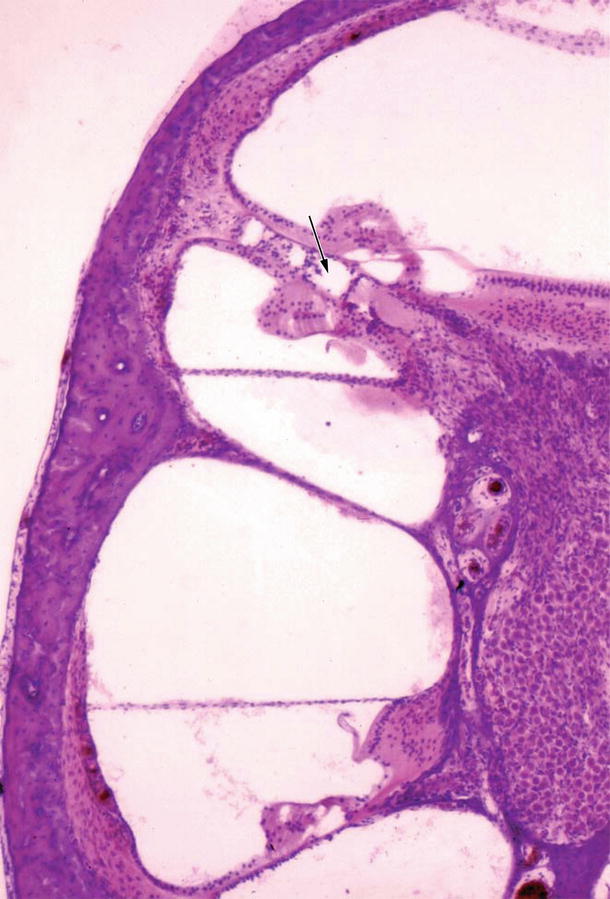
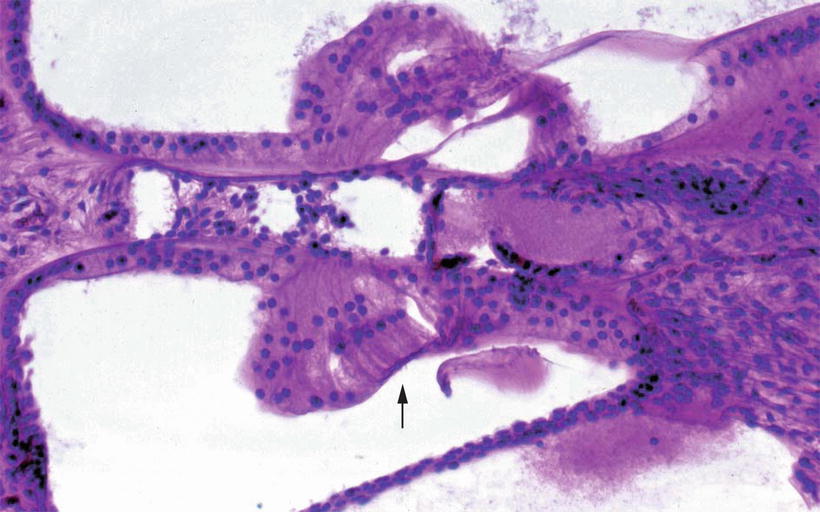
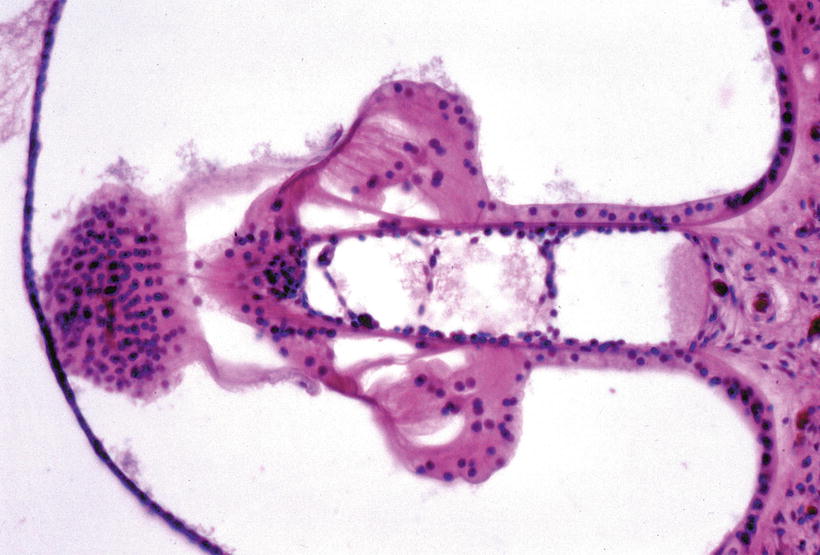
Fig. 9.1
Graphic reconstruction of the cochlear coils showing hyperplasia of the organ of Corti. Of a total length of 24 mm, the excessive organ of Corti occupies 1 mm [2]
Fig. 9.2
Mirror image of the organ of Corti [2]. Arrow indicates the common narrow scala tympani. The inverted scala media together with the scala vestibuli lies in the space normally occupied by the scala tympani. Nerve fibers are fewer in number toward the inverted organ of Corti (original ×2.5)
Fig. 9.3
Higher power view of Fig. 9.2. Excessive outer hair cells are present (arrow). The inner sulcus is absent. The osseous spiral lamina and nerve fibers are hypoplastic. The inverted limbus is not well developed. The detached tectorial membrane shows small projections toward the limbus. The stria vascularis is atrophic (original ×16)
Fig. 9.4
Mirror image of the organ of Corti. Deiters’ cells beneath the outer hair cells are partly missing. Cellular bridges in the common scala tympani connect the upper and lower tympanic lamella. No nerve fibers are observed in the organ of Corti (original ×16) [2]
Merchant and Nadol [3] reported a similar case from the human temporal bone collection at the University of Zurich. The bone came from an 18-year-old man with congenital syphilis, who died of unknown causes. In his left ear, the organ of Corti of the shortened apical turn was inverted so that its cuticular plate faced the cuticular plate of the second turn. The limbus spiralis was present. The normal and inverted portions shared a common tectorial membrane. Hair cells were present. The stria vascularis was hypoplastic in the region of the anomaly. In this case, the inverted organ of Corti shared the scala media of the second turn, whereas in the guinea pig, the scala media itself was inverted, together with all other constituents.
Some viruses are known to cause anomalous development of the inner ear. These include Herpesviridae, Togaviridae, and Bunyaviridae. A virus may attack the anlage, killing or degenerating part of it. Because the anlage is the first step of organ formation, cessation of cell division or decreased rate of cell division occur and cause further changes in the anlage. Malformations are usually due to cessation of development or partial loss of the organ.
In contrast to the usual developmental anomalies, the cochlea described above is characterized by excessive development of the cochlear duct, which returned at the apex with the scala vestibuli down into the space normally occupied by the scala tympani. Perhaps in this case the signals to create the normal organ of Corti were not turned off, resulting in its hyperplasia. Whether or not GPCMV induced mutation in the gamete is uncertain.
9.2 Hypophosphatasia
9.2.1 Introduction
Hypophosphatasia, named by Rathbun in 1948 [4], is a rare genetically determined metabolic disease characterized by defective bone and tooth mineralization. The disease results from mutations in the liver/bone/kidney alkaline phosphatase gene encoding tissue-nonspecific alkaline phosphatase. The diagnosis is based on laboratory assays and DNA sequencing of the ALPL gene [5].
Hypophosphatasia is characterized by (1) abnormal mineralization of bone (2) lowered serum and tissue alkaline phosphatase activity, and (3) increased urinary excretion of phosphoethanolamine. Age of onset varies considerably from before birth to adulthood. The earlier the onset of pathology, the more severe the lesions. There is no curative treatment for hypophosphatasia.
Fraser [6] divided hypophosphatasia cases into three groups, based on the estimated time of development of demonstrable bone lesions:
Group 1:
Infants with lesions present at birth or within the first 6 months of life
Group 2:
Children with lesions gradually becoming apparent after the age of 6 months
Group 3:
Individuals with lesions appearing in adulthood
9.2.2 Case Report
9.2.2.1 Clinical Findings
Nomura and Mori [7] reported a case of hypophosphatasia belonging to Group 1 of Fraser’s classification.
A full term girl was born to consanguineous parents. The delivery was normal. The baby weighed 2,140 g at birth and died 1 h later. Her 29-year-old mother had no obvious disturbances in the course of pregnancy. Examinations had indicated an uneventful pregnancy. The fetal outline, however, could not be identified and radiography had failed to show the skull and skeleton.
Autopsy revealed the following findings:
1.
The baby had stunted extremities: arms and legs measured 5 cm and 6 cm, respectively. No malformations were found in the face, head, or neck.
2.
Bones were soft. Histology found inhibited calcification of the cartilaginous bone. The epiphyses were irregular and thick. These findings were similar to those of rickets.
3.
The skull was peculiarly soft.
4.
There was enamel hypoplasia of the deciduous teeth.
5.
The scapulae were partly ossified.
6.
The lungs showed marked dystelectasis.
Serum alkaline phosphatase could not be measured because of the infant’s early death. The enzyme levels were low in her parents (father 1.5 units/100 mL, mother 2.0 units/100 mL, normal 3–13 King-Armstrong units/100 mL).
9.2.2.2 Histopathology of the Temporal Bones
Temporal bones were removed and submitted for histopathological study. The bones were easily severed from the skull with scissors. Decalcification was not necessary for temporal bone sectioning. The specimens were embedded in celloidin and sliced into serial sections 20 μm thick.
The Middle Ear
The malleus, incus, and stapes were present as usual in the middle ear cavity, which was occupied by mesenchymal tissue. The ossicles were osteoid. Bone marrow was also present.
The Inner Ear
The otic capsule was composed of red (eosin) osteoid tissue and bone marrow. The membranous labyrinth of the pars superior and pars inferior was well developed. The organ of Corti appeared fairly normal. The populations of hair cells and ganglion cells were normal.
The spiral ligament showed a peculiar change. A portion of the spiral ligament underlying the stria vascularis was densely stained blue by hematoxylin (Fig. 9.5). This seemed to be calcified tissue, with an occasional blood vessel running through it (Fig. 9.6). The tissue was continuous and most striking from the upper basal turn to the upper middle turn.
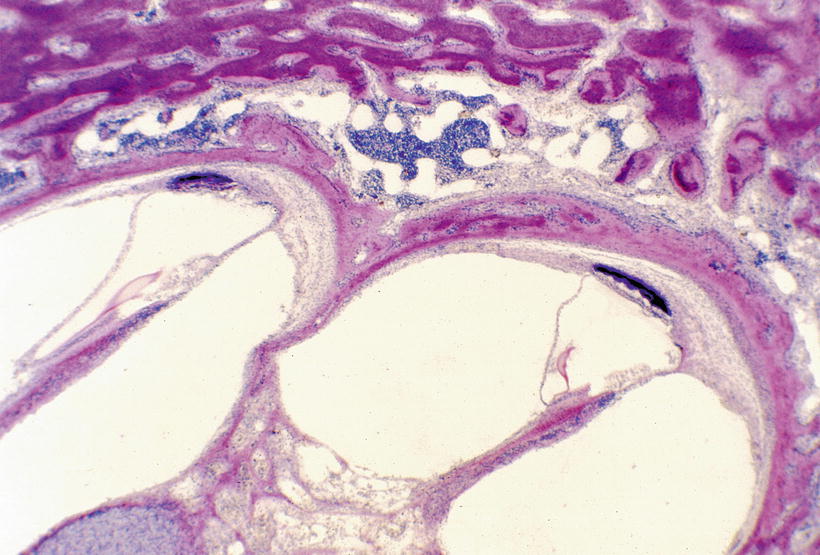
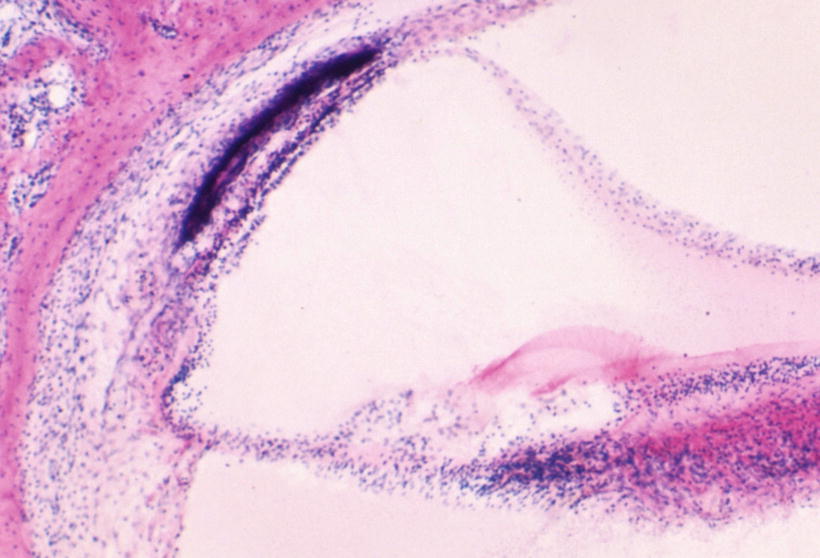
Fig. 9.5
Temporal bone from a patient with hypophosphatasia [7]. Between the stria vascularis and the spiral ligament, there is tissue densely stained by haematoxylin. The otic capsule is red-eosinophilic osteoid. The organ of Corti and dendrites in the osseous spiral lamina are normal. HE staining (original ×2.5)
Fig. 9.6
High power view of the stria vascularis and spiral ligament. The stria vascularis is thin. The densely stained tissue is behind, but not directly bordering, the stria. There is hypocelluar area between them. A capillary runs through the densely stained tissue. The spiral ligament contains a hypocellular zone. HE staining (original ×6.5)
In the left cochlea the densely staining tissue was continuous for 9.4 mm from the 10 mm area of the basal turn. In the right ear the tissue extended for 9.6 mm from the 8 mm area. The total lengths of the left and right cochleae were 28.5 and 28.3 mm, respectively. The tissue lay beneath the stria vascularis, however, an amorphous tissue zone was present between the stria and the blue haematoxylin tissue. Various staining methods were employed, revealing the tissue to be PAS positive, blue by Azan, Hale’s colloid iron negative, and with no metachromasia by toluidine blue. This material was interpreted to be either calcification or hyaline degeneration. Varying amounts of amorphous substance existed between this tissue and the stria vascularis (Fig. 9.7).
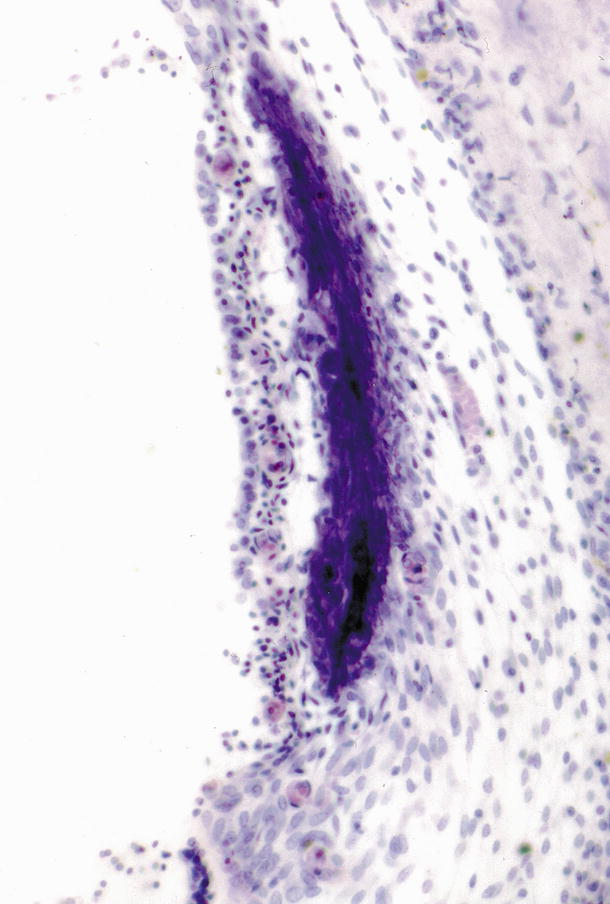
Fig. 9.7
High power view of the densely stained tissue [7]. There is a lucent area between the stria vascularis and the blue-stained tissue. The stria vascularis and spiral prominence seem underdeveloped, although several capillaries are observed in cross-section. The spiral ligament is hypocellular. HE staining (original ×16)
Hypophosphatasia is caused by mutations in the ALPL gene. This gene codes for an enzyme called alkaline phosphatase, which plays an essential role in bone and tooth mineralization. Mutations in the ALPL gene lead to the production of an abnormal version of alkaline phosphatase, which cannot function in the normal process of mineralization.
It is generally thought that elevation of serum alkaline phosphatase activity in the absence of liver disease reflects osteoblastic activity. Enzyme levels are highest during the most rapid growth phase and become abnormally elevated when osteoblast activity increases in such bone diseases as rickets, hyperparathyroidism, and Paget’s disease. Phosphoethanolamine is considered the true substrate for alkaline phosphatase.
Although serum alkaline phosphatase and phosphoethanolamine levels were not determined in this case, the following factors made it possible to diagnose hypophosphatasia: (1) abnormal mineralization of bone as evidenced by radiography (2) histologically-demonstrated bony changes similar to rickets, though congenital rickets was ruled out, and (3) consanguineous parents showing low levels of serum alkaline phosphatase activity.
The histologic lesions of hypophosphatasia are characterized by two abnormalities [6]: (1) disturbance in the normal calcification process of cartilage and bone, (2) excessive production and disordered arrangement of preosseous cartilage and of osteoid. It has been stressed that the lesions of hypophosphatasia are morphologically indistinguishable from those of simple rickets unless special stains are performed to demonstrate phosphatase.
As far as the otic capsule is concerned, typical changes in rickets are confined to the periosteal layer with newly formed bone lamellae. In marrow spaces the newly formed bone lamellae are poorly calcified and show a red-eosinophilic osteoid border. Osteoid is the unmineralized, organized portion of the bone matrix. The endochondral layer remains normal unless active bone resorption occurs. The inner ear remains normal.
The PAS positive tissue in the spiral ligament in this case is strange and might be pathognomonic of hypophosphatasia. PAS positive material in the stria vascularis has occasionally been reported [8–11] and has been interpreted as hyaline cartilage or calcification. The present case did not show such changes in the stria. The PAS positive tissue in the spiral ligament was not hyaline, but was possibly calcified tissue.
Dr. Schuknecht examined the specimen and observed that the region of the spiral ligament underlying the stria vascularis showed changes consistent with strial atrophy. Rauch [12] described strong alkaline phosphatase activity in the spiral ligament of guinea pig embryos. His histochemical study demonstrated that the enzyme was abundant in this particular perilymph-endolymph junction area. The finding of abnormal tissue in the spiral ligament was difficult to explain in relation to alkaline phosphatase. This tissue may be comparable to pseudocalcinosis of the brain.
9.3 Alagille Syndrome
9.3.1 Phylogenesis of the Semicircular Canal
Phylogenetically, a single semicircular canal appeared in the hagfish (Myxinoidea), an eel-like animal, during the Cambrian Period some 530 million years ago (Fig. 9.8) [13]. The single canal has two ampullae and a macula communis. It is in the hagfish that the first true vestibular apparatus developed.
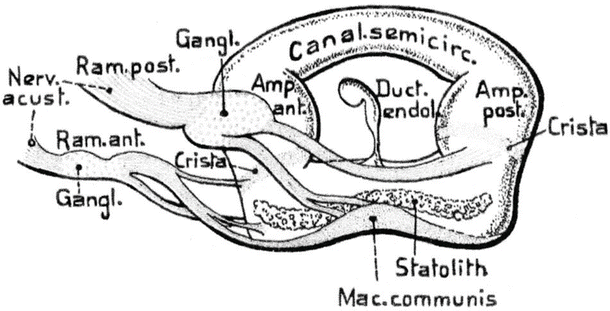
The lamprey (Petromyzontida) shows higher development of the inner ear, with two vertical canals and three ampullae. The vestibulum has become more complex than in the hagfish (Fig. 9.9a, b). The hagfish and lamprey belong to the order Cyclostomata. The former are found only in salt water, and the latter in both salt and fresh water. Both have cartilaginous capsules.
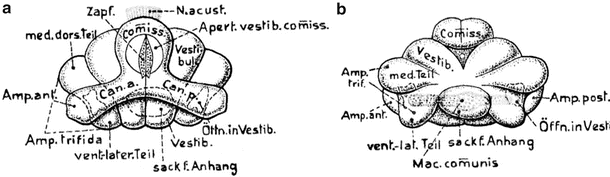
Fig. 9.9
Lamprey—left membranous labyrinth. (a) Dorsal view, (b) ventral view. From Bütschli after Retzius [13]
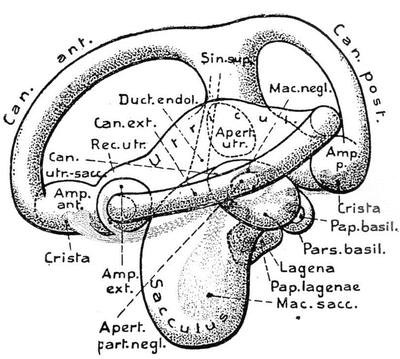
Three semicircular canals appeared in fish that must have developed in the Devonian Period of the Paleozoic Era, about 350 million years ago. The Japanese eel (Anguilla japonica) has three canals. The general arrangement of the membranous labyrinth in all bony fishes is very similar. An extremely large utricule is found in the lungfish (Protopterus annectens) (Fig. 9.10). In the sturgeon (Acipenser sturio) the recessus utriculi is much smaller, as is the saccule. The European perch (Perca fluviatilis) possesses an elongated sinus superior utriculi, a small main utricular body, and a large saccule (Fig. 9.11). The sinus superior utriculi is a part of the utricle, and a predecessor of the crus commune. The lateral canal appeared later and is shorter than the two vertical canals [14].
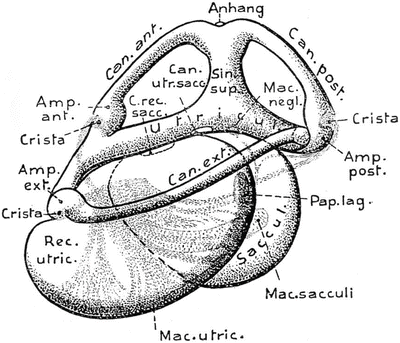
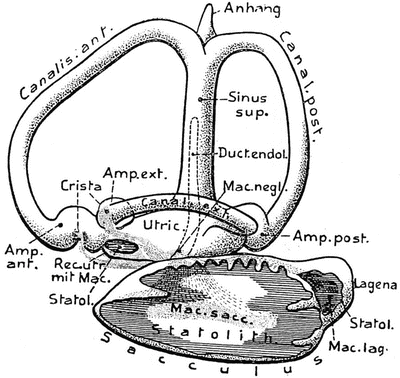
Fig. 9.10
Protopterus annectens―lungfish Left labyrinth, lateral view From Bütschli after Retzius [13]
Fig. 9.11
Perca fluviatilis―European perch. Left labyrinth, lateral view Mac. negl., macula neglecta. From Bütschli after Retzius [13]
The membranous labyrinth of the frog (amphibian) and dove (avian) are shown in Figs. 9.12 and 9.13. The hearing organ begins its evolution. The pars basilaris makes its appearance in the amphibian ear. In birds the pars basilaris has elongated as the membrana basilaris, which becomes so extensive in mammals that it needs to form a spiral to find space. The very ancient balance organ reached perfection in primitive animals, whereas the hearing organ has continuously evolved along with mammals, keeping pace with the needs of the various life forms [14].