Purpose
To present the current understanding of age-related macular degeneration (AMD) pathogenesis, based on clinical evidence, epidemiologic data, histopathologic examination, and genetic data; to provide an update on current and emerging therapies; and to propose an integrated model of the pathogenesis of AMD.
Design
Review of published clinical and experimental studies.
Methods
Analysis and synthesis of clinical and experimental data.
Results
We are closer to a complete understanding of the pathogenesis of AMD, having progressed from clinical observations to epidemiologic observations and clinical pathologic correlation. More recently, modern genetic and genomic studies have facilitated the exploration of molecular pathways. It seems that AMD is a complex disease that results from the interaction of genetic susceptibility with aging and environmental factors. Disease progression also seems to be driven by a combination of genetic and environmental factors.
Conclusions
Therapies based on pathophysiologic features have changed the paradigm for treating neovascular AMD. With improved understanding of the underlying genetic susceptibility, we can identify targets to halt early disease and to prevent progression and vision loss.
It is an honor to deliver the LXIX (69th) Edward Jackson Memorial Lecture. Dr Jackson was drawn to ophthalmology after diphtheria paralyzed his accommodation. In his career, he made practical contributions in physiologic optics, including popularizing retinoscopy and developing the Jackson cross cylinder, but was also interested in ocular infections and inflammation. He was dedicated to the education and training of ophthalmologists, and it is therefore fitting to honor him with an annual lecture at the American Academy of Ophthalmology. It is a privilege to add my name to the list of Jackson lecturers, which includes some of the greatest names in ophthalmology past and present. Note that I am only the second woman to be so honored.
Today I will be discussing age-related macular degeneration (AMD), a subject that was last presented by Dr Stuart Fine in his 2004 Jackson Lecture. AMD occurs in neovascular (exudative or wet) and nonneovascular (nonexudative or dry) forms, and according to the World Health Organization, it is the leading cause of blindness in developed countries and ranks third worldwide after cataract and glaucoma. At the time that Dr Fine presented his lecture, laser photocoagulation was still used to treat neovascular AMD; however, its use was diminishing with the approval of verteporfin photodynamic therapy (Visudyne; Quadra Logic Technologies Inc, Vancouver, Canada), and the field was on the verge of innovations for neovascular AMD.
Now in 2012, retina specialists are comfortable, although not completely satisfied, with the results we obtain in patients with neovascular AMD. Treatments targeting vascular endothelial growth factor (VEGF) have been shown to halt vision loss in more than 90% of patients and to improve vision in one third. Despite these successes, there is a natural desire to achieve even better results and to reduce treatment burden. Moreover, because the available treatments largely are aimed at late stages of AMD, we need to be able to intervene in the early stages of the disease so we can forestall progression. In 2012, we are on the verge of developing treatments that will prevent the disease and preserve normal visual function. Using a combination of genetic and functional studies, we will have a better understanding of the pathogenesis of AMD, which will allow us to design more elegant treatments and to achieve success in prevention and early intervention.
The goal of this lecture is not to provide a comprehensive overview of AMD; readers are directed to reviews by Jager and associates and de Jong and associates. Rather, the goal is to review the evolution in our understanding of AMD pathogenesis and attempt to formulate an integrated model of AMD. To this end, I briefly review the natural history of AMD, then review aspects of pathogenesis that have been elucidated over the years—particularly aging, lipid and lipoprotein deposition, inflammation (including complement), angiogenesis, and cell death pathways. Next, I discuss the recent explosion of genetic information that has provided new insights into AMD pathogenesis, in some cases supporting previous observations, and in others offering unexpected clues. Treatment approaches—past, current, and in development—are discussed in a supplemental appendix . Finally, I propose a comprehensive model of AMD, incorporating clinical observations, molecular evidence, and genetic discoveries.
The Natural History of Age-Related Macular Degeneration
The earliest clinical manifestations of AMD are drusen and pigmentary changes in the macula ( Figure 1 , Top row). Drusen are focal deposits of extracellular debris that typically form between the retinal pigment epithelium (RPE) and Bruch membrane, and are described clinically by their size and contour. Small drusen (<63 μm) are not indicative of AMD and can be seen in the eyes of healthy young and middle-aged adults. Intermediate drusen (63 to 125 μm) and large drusen (>125 μm) are characteristic of AMD when located within the macular region ( Figure 1 , Top middle). Large, soft drusen typically have indistinct borders and, when large enough, are really focal RPE detachments. Cuticular or basal laminar drusen, which are not believed to be associated with aging or AMD, appear as a multitude of sharp-bordered subretinal lesions; these are small, uniformly sized, yellow, and most evident on an infrared image. In contrast, subretinal drusenoid deposits, or reticular pseudodrusen, are associated with AMD and are believed to be predictive markers for progression to advanced AMD. Reticular pseudodrusen appear as a network of yellow, oval, or roundish lesions with a diameter of 125 to 250 μm. As shown in Figure 1 (Middle row), they are best seen with red-free imaging or infrared wavelengths of the scanning laser ophthalmoscope, and are not hyperfluorescent on fluorescein angiography. Early explanations attributed these lesions to choroidal vascular nonperfusion, but spectral-domain optical coherence tomography (OCT) indicates that the lesions are anterior to the RPE and extend upward into the photoreceptor inner and outer segments ( Figure 1 , Middle row center). Focal hyperpigmentation of the RPE, a common finding in AMD, appears as focal areas of grey or black pigment ( Figure 1 , Top right). Drusen and pigmentary changes alone do not always compromise central visual acuity, although other visual function abnormalities may occur.
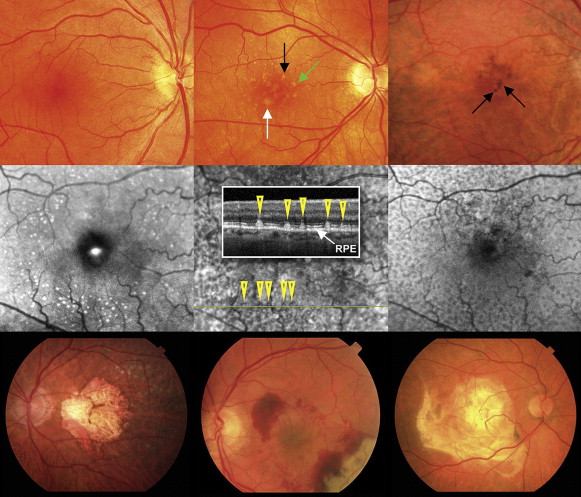
The progression of AMD can be identified by clinical examination of the macula. Classification schemes based on macular features provide the most reliable prognostic information currently available, and phenotypes based on these classification schemes are independent variables in risk assessment models for advanced AMD. The Age-Related Eye Disease Study (AREDS) 9-step severity scale and the simplified 5-step severity scale use drusen size and pigmentary abnormalities to determine a risk score ( Table 1 ).
| Drusen Classification a | Size (μm) | Risk Factor Scoring b | |||
|---|---|---|---|---|---|
| Small | <63 | +1: for each eye with large drusen | |||
| Intermediate | 63 to 124 | +1: for each eye with pigment abnormalities | |||
| Large | 125 to 249 | +1: if neither eye has large drusen, intermediate drusen in both eyes | |||
| Very large | >250 | +2: for the eye that has neovascular AMD | |||
| Approximate 5-year rates of progression to advanced age-related macular degeneration b | |||||
| Score | 0 | 1 | 2 | 3 | 4 |
| Risk | 0.5% | 3% | 12% | 25% | 50% |
a Adapted from Age-Related Eye Disease Study report no. 17.
Although drusen are the first clinically visible abnormality in AMD, they may not be the first detectable change. Impaired dark adaptation upon moving from brightly lit to dim environments is commonly reported by patients with early AMD, although central visual acuity remains normal. It has been observed recently that abnormalities in dark adaptation may precede clinically detectable macular changes. This suggests that rod photoreceptor dysfunction is an early manifestation of the disease. Testing dark adaptation thus may prove to be a useful screening tool as well as a surrogate outcome for clinical trials in early AMD.
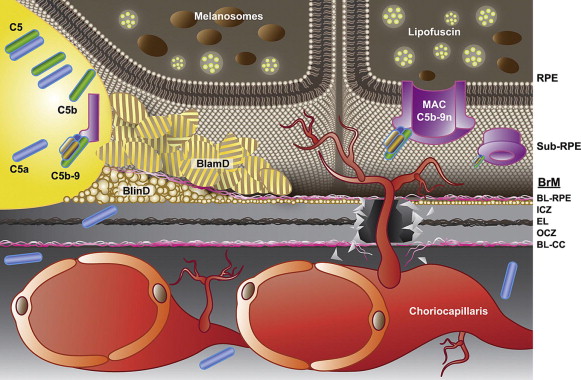
Pathologically, alterations of the RPE and Bruch membrane characterize the early changes of AMD. The RPE seems to be the site of initial cellular injury in AMD, perhaps because of changes in the Bruch membrane (described below), as well as the accumulation of lipoprotein (including lipofuscin) in the RPE itself. The RPE cellular responses to injury likely mediate the pathologic processes involved in disease establishment and progression. The Bruch membrane is an extracellular matrix (ECM) consisting of 5 layers: the basement membrane (or basal lamina) of the choriocapillaris, the outer collagenous zone, the central elastin layer, the inner collagenous zone, and the basal lamina of the RPE ( Figure 2 ). The Bruch membrane undergoes constant turnover, mediated by the matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs). Elastin previously was thought to remain stable, although more recent studies have indicated that elastic fibers in the ECM do in fact turn over, and homeostasis is dependent on the lysyl oxidase-like 1 protein, which interacts with fibulin 5 and colocalizes with sites of elastin formation. Defects in elastin metabolism very well may affect the Bruch membrane in AMD. This is evidenced by the observation that eyes with AMD exhibit larger gaps in macular elastin layer integrity compared with controls. The Bruch membrane thickens progressively with age, partly because of increased levels of TIMPs and a resulting reduction in ECM turnover. The accumulation of lipid-rich basal laminar deposits (BlamD) and basal linear deposits (BlinD) also contribute significantly to thickening of the Bruch membrane ( Figure 2 ). The terms BlamD and BlinD have been defined variably by different authors, and their definition may depend on whether one describes light or electron microscopic findings. Both BlinD and BlamD are considered basal deposits. Curcio and associates have described BlinD as a layer of membrane-bound vesicles, often external to RPE basal lamina that can be continuous with soft drusen. They observed that BlinD and soft drusen have the same composition, and therefore are likely to represent diffuse and focal deposits of the same material. In contrast, BlamD consist primarily of fibrous long-spacing collagen and amorphous material, and occur with both aging and AMD.
Advanced, late stages of AMD are characterized by geographic atrophy (GA), choroidal neovascularization (CNV), or both, as shown in Figure 1 (Bottom row). GA can manifest as single or multiple areas measuring 175 μm or more of RPE loss or depigmentation, with associated choriocapillaris atrophy. GA is evident on OCT as a loss of the corresponding retinal layers and on fundus autofluorescence as loss of the normal autofluorescence in the area. Based on the increased hyperfluorescence at the junctional zone of atrophy, the Fundus Autofluorescence in Age-Related Macular Degeneration study group classified GA lesions into 4 primary phenotypes: focal (single or individual small spots of increased fundus autofluorescence (FAF) at the margin of the atrophic patch), banded (continuous stippled-band or ring-shaped zone of increased FAF surrounding the entire atrophic area), patchy (large patchy areas of increased FAF outside the atrophic area), and diffuse (increased FAF at the margin of the atrophic area and beyond). Areas of GA correspond with loss of visual function on microperimetry.
Neovascular AMD is characterized by neovascularization arising from the choroid into the subretinal (type 1) or sub-RPE (type 2) space. Some controversy exists as to whether the vessels in retinal angiomatous proliferation, a neovascular phenotype, arise from the choroid or retina. Indeed, both may occur; thus, Freund and associates proposed a type 3 classification of intraretinal neovascularization to denote intraretinal vascular complexes that arise from both deep retinal capillaries and the choroid. Polypoidal choroidal vasculopathy (PCV), originally described as a maculopathy distinct from AMD, also has been shown to occur as a form of neovascular AMD and is characterized by grape-like clusters of new vessels (usually in the sub-RPE space). PCV is the more common presentation of neovascular AMD in populations of Asian or African descent. Symptoms in neovascular AMD relate to elevation of the retinal layers with associated metamorphopsia or vision loss when retinal layers are compromised by blood, fibrosis, and cell loss. In eyes with AMD, subretinal fluid usually indicates neovascularization and should be investigated with OCT and angiography. However, subretinal fluid also can occur in the absence of neovascularization (confirmed by fluorescein and indocyanine green angiography). In these atypical cases, subretinal fluid most likely occurs because of dysfunction of the RPE outer retinal barrier.
Pathogenesis: An Evolving Story
Our understanding of AMD and its pathogenesis has changed over decades of observation. Although there has been progress, there are still missing pieces in the puzzle, and any attempt at a unified model will meet with disagreement. Nevertheless, formulating a model is a worthwhile exercise and a prerequisite to developing biologically based therapies. However, before developing a model based on current knowledge, it is useful to review the pathogenesis of AMD from a historical perspective.
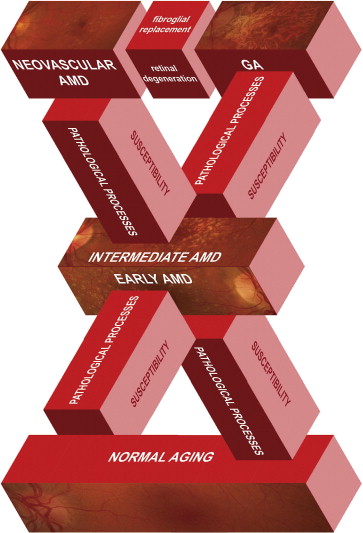
Senile macular degeneration first was described as a clinical disorder by Haab in 1885 (reviewed by Gass ), but for decades, clinicians did not make the connection between macular drusen and disciform scars, which represent the end stage of exudative AMD ( Figure 1 , Bottom right). J. Donald Gass, the keen observer here as he was for so many retinal entities, described the progression of macular drusen to serous RPE, retinal detachments, and neovascularization in the subretinal and sub-RPE space, as well as describing GA. Many of Gass’ early observations remain clinically relevant. Patients with extramacular or peripheral drusen, or both, sparing the macula rarely lose vision, and macular drusen often disappear with the onset of serous detachments or neovascularization. Gass also noted that the disorder was more common in families, supporting a genetic contribution, and he believed that GA and neovascularization were different manifestations of the same disease. Although some have questioned whether the different phenotypes represent different diseases, Gass’ conclusion—that all are related forms of AMD—is probably correct. Figure 3 illustrates this concept and emphasizes the interconnections of AMD stages.
Epidemiologic studies, including the Framingham Eye study, First National Health and Nutrition Examination Survey, and the Eye Disease Case-Control Study, sought to understand the pathogenesis of AMD by evaluating risk factors. Advanced age is the greatest risk factor for AMD, and cigarette smoking remains the most consistent preventable risk factor across all studies. Associations with cardiovascular risk factors, although varying individually, are consistent as a group; for instance, hypertension was found to be associated in some studies, whereas elevated cholesterol was associated in others. Although an association has been postulated between ultraviolet light exposure and AMD, the Eye Disease Case-Control Study found no association. This was in agreement with previous studies by West and associates, who examined a cohort of Chesapeake Bay watermen and found no association between ultraviolet B or ultraviolet A exposure and AMD. However, Taylor and associates did find an association between visible light exposure and advanced AMD (GA or neovascular AMD). Hyperopia was identified as a risk factor for CNV in a 1993 study conducted at Massachusetts Eye and Ear Infirmary, a finding that was corroborated by several large studies–notably AREDS, the Rotterdam Study, and the Beijing Eye Study. It has been suggested that hyperopic shortening may lead to abnormal choroidal perfusion and increased choroidal resistance, thus predisposing eyes with shorter axial lengths to AMD. Higher serum levels of carotenoids, large horizontal cup-to-disc ratios, and postmenopausal estrogen use in women were associated with a reduced risk of neovascular AMD. Risk factors that have been suggested variably but are no longer a subject of focus include left ventricular hypertrophy, decreased hand grip strength, short height, decreased vital capacity, and history of lung infection, as well as parity greater than zero for neovascular AMD.
Clinical observations and epidemiologic data stimulated the development of mechanistic models of AMD pathogenesis. Further clinical pathologic correlations and biochemical investigations contributed to these ideas. The changes observed in early AMD are now known to involve many physiologic processes that may become pathologic over time and may be exacerbated by genetic or environmental factors.
Age and Lipid Deposition
Age is the greatest risk factor for AMD, and even with normal aging, the Bruch membrane undergoes neutral lipid deposition, as demonstrated by Pauleikhoff and associates in 1990. The notion of lipid accumulation as an early harbinger of AMD remains a current concept and invokes comparisons with atherosclerosis, another age-related disease with both hemodynamic and lipid-based hypotheses. However, the lipid-rich lesions (such as BlinD and drusen) are distinctive to AMD and do not necessarily reflect an aging process. Thus, understanding what is different about lipid accumulation in AMD should provide insight into the pathogenesis of early AMD.
On the basis of its binding with oil red-O, neutral lipid was identified in drusen in the 1960s, and emerging evidence suggests that lipid accumulation has an underlying role in the pathogenesis of AMD. Studying the hydrodynamics of the Bruch membrane using donor tissues, Starita and associates demonstrated that hydraulic conductivity declines with age, and that impaired transport resulting from lipid deposition may contribute to AMD. More recently, it has become apparent that deposits in the Bruch membrane likely arise from multiple sources, including outer segments debris, systemic circulation, and endogenous synthesis. Having demonstrated through accumulated evidence that RPE is the likely source for lipoprotein accumulation in both aging and in AMD, Curcio and associates formulated a compelling model for AMD pathogenesis. According to their model, the Bruch membrane functions much like the endothelium in atherosclerosis, and parallel processes in AMD and atherosclerosis can be traced to lipid deposition. Specifically, lipoproteins such as apolipoprotein B, which serve the biological function of delivering cholesterol to tissues, become retained in the subendothelium of arteries. Oxidative and nonoxidative modifications subsequently trigger pathologic adaptive responses. This model of atherosclerosis, known as the response-to-retention hypothesis, provides a feasible explanation for comparable processes in AMD: apolipoprotein B and other lipids (likely produced by the RPE) accumulate in the Bruch membrane and sub-RPE space, and pathologic responses to these retained particles lead to AMD. Thus, whereas atherosclerosis is the consequence for relying on apolipoprotein B for cholesterol and lipid transport, AMD is the consequence for relying on similar systems to transport fat-soluble nutrients (such as retinol) in the retina.
Examining age-related changes in human eyes using quick-freeze or deep-etch techniques, Ruberti and associates observed the accumulation of esterified cholesterol and unesterified cholesterol in the Bruch membrane. They showed that in early adulthood, esterified cholesterol- and unesterified cholesterol-containing particles form a thin sublayer; by 60 years of age, a confluent, hydrophobic barrier forms, with the esterified cholesterol and unesterified cholesterol concentrated in macular regions. Because the observed lipid wall coincides with the location of BlinD (see Figure 2 ), which are characteristic of early AMD, it is likely that the lipid wall precedes the formation of BlinD and drusen. Further bolstering this model, Curcio and associates showed that drusen are composed of at least 40% lipid. These observations support the hypothesis that AMD, like atherosclerosis, has a pathologic component that involves lipid deposition.
Photoreceptor distribution also changes with age. The macula is characterized by a cone-dominated fovea surrounded by a rod-dominated parafovea. Cones are stable in adulthood, but rods decrease by 30%, with even greater loss overlying sub-RPE deposits. Interestingly, as the rods die, the surviving rod inner segments expand to fill the vacated space, suggesting that the rods and cones actively regulate their inner segment space allocation. RPE cell morphologic features also show topographical and age-related differences. Early in life, the RPE cells of the macular area are broader and lower, becoming narrower and higher toward the equator and periphery. In older eyes, this gradient is reversed: cells in the macular area become narrower and higher, whereas cells in the periphery become broader and lower with little change in the equatorial zone.
Vascular Resistance
Based on the similarities between AMD and atherosclerosis, Friedman postulated that hemodynamic changes and increased choroidal resistance underlie the pathogenesis of AMD. He traced the hypothesis back to Friedenwald’s observation that scleral rigidity increased with age, and to Verhoeff and Grossman’s observation that AMD seemed to be associated with drusen and impaired choroidal flow. Friedman showed that increased scleral rigidity was associated with AMD compared with age-matched controls and attributed this increase in scleral rigidity to lipid deposition—similar to atherosclerosis. He had observed that drusen appeared in the posterior pole and adjacent to vortex veins anteriorly, where the venous pressure is greatest, suggesting that their formation relates in part to a venous abnormality. Friedman postulated that progressive infiltration by lipids in the sclera and other ocular tissues leads to increased resistance in the choroidal vessels, thereby diminishing the ability of the choriocapillaris to clear lipoproteins from the RPE and Bruch membrane. Hyperopia, a risk factor previously identified, would contribute to increased resistance in choroidal vessels, and thus supports the hemodynamic model of AMD. Choriocapillaris density also has been shown to be increased significantly in both neovascular and nonneovascular AMD compared with controls, suggesting increased resistance and impaired flow. As a consequence of these changes, metabolic exchange across the RPE may be compromised, and lipoproteins from photoreceptors and other sources could accumulate in the Bruch membrane as well as the outer retina to form drusen. In proximity to the RPE, these alterations could lead to atrophy; additionally, by altering the Bruch membrane with subsequent calcification and fracture, the accumulation could lead to neovascularization.
Lipofuscin Accumulation
Light-sensitive photoreceptor outer segments are renewed constantly, and the RPE functions to phagocytose photoreceptor outer segment discs for lysosomal degradation. In the 1970s, Hogan surmised that age-related changes in the Bruch membrane and the RPE result from the accumulation of by-products of this phagocytic process. Lipofuscin, a granular pigment that results from lysosomal digestion, has been shown to accumulate in the RPE with age. Lipofuscin accumulation in the RPE results in increased fundus autofluorescence, which has been suggested to be a prelude to GA. Lipofuscin is not defined completely, but several fluorophores have been isolated. One important fluorophore is A2E, a pyridinium bis-retinoid composed of 2 vitamin A aldehyde molecules and 1 ethanolamine molecule. A2E accounts for the fluorescent properties of lipofuscin and forms the basis of clinical fundus autofluorescence imaging. A2E has been shown to inhibit lysosomal degradative function and cholesterol metabolism in the RPE. A2E is capable of acting as a photosensitizer and has been shown to mediate blue-light-induced apoptosis. Furthermore, A2E has been demonstrated to induce the complement system of innate immunity, which has an emerging role in AMD pathogenesis (discussed below). Via these mechanisms, A2E may have toxic effects on the RPE, leading to cell death and atrophy. In murine models of macular degeneration and other retinal degenerative disorders, lipofuscin accumulation often is associated with neural degeneration (reviewed in Ramkumar and associates ). Although lipofuscin accumulation is central to the pathologic process, determining whether this is cause or effect is critical to understanding the initiation of AMD.
Amyloid Accumulation
Several studies have shown that similar processes cause drusen formation in the eye and senile plaque formation in the brain, invoking comparisons between AMD and another age-related condition: Alzheimer disease. Both diseases exhibit the accumulation of extracellular deposits such as apolipoprotein E (ApoE) and amyloid-beta (Aβ). ApoE functions in normal lipid catabolism and transport and is a major lipoprotein component of drusen. Variants of the APOE gene have been implicated in Alzheimer disease as well as AMD, although the risk associations are in opposite directions. The E4 allele is a risk factor for Alzheimer disease, but seems to be protective for AMD, whereas the E2 allele is a risk factor for AMD. Aβ, a peptide that forms a large proportion of senile plaques, has been identified as a significant component in drusen and has been implicated in inflammatory events (discussed below) that contribute to RPE dysfunction in AMD. Subsequent studies have shown that Aβ-containing drusen are specific to the retinas of AMD patients and seem to correlate with advanced stages of AMD. There may be a genetic link between AMD and Alzheimer disease in the amyloid pathway, because a gene variant of cystatin C (believed to be involved in amyloid deposition) has been linked to increased susceptibility to both diseases. Using an antibody to target Aβ peptides in the APOE4-HFC mouse model of AMD, Marcu and associates demonstrated reduced levels of both Aβ and activated complement components in sub-RPE deposits and structural preservation of the RPE. According to in vitro studies, AMD-related Aβ is likely of RPE origin and is proangiogenic, inducing VEGF expression while decreasing the expression of pigment epithelium-derived factor (PEDF), an endogenous antiangiogenic protein. Interestingly, Aβ recently was demonstrated to accumulate in photoreceptor outer segments as an age-related change in normal human retinas, suggesting that it may be associated with visual function at the level of the neural retina. Although AMD and Alzheimer disease both exhibit apolipoprotein and amyloid deposition, several differences remain, including the disparity of genetic risk factors. Moreover, just as the amyloid hypothesis for Alzheimer disease has been questioned, it remains unclear whether Aβ plays a causative role in AMD.
Inflammation and Immune Response
In addition to age-related changes such as hemodynamic alterations and lipid accumulation, inflammation is recognized widely as another important player in the pathogenesis of both atherosclerosis and AMD. Genetic variation in the complement pathway (discussed below) and environmental factors (like cigarette smoking) may predispose individuals to the disease through inflammatory mechanisms. Moreover, inflammatory responses can upregulate VEGF and other proangiogenic factors that are essential for CNV, linking inflammation with the progression of AMD to advanced stages.
Long before the link between AMD and complement gene variants became widely recognized, Hageman and associates were vigorous proponents of a role for inflammation in the pathogenesis of AMD, based on observations of human specimens. Hageman and associates postulated that inflammatory or immune-mediated processes (including the recruitment and activation of dendritic cells) were central to the development of drusen and subsequent events. This notion is supported by clinicopathologic studies: multinucleated giant cells, mononuclear phagocyte system (MPS) cells, various immunocompetent cells, and increased HLA-DR immunoreactivity in retinal microglia have been described in histopathologic specimens from AMD patients. Age-related changes in retinal microglial activation, which have been linked to lipofuscin accumulation, may contribute to immune dysfunction in AMD pathogenesis. Infectious agents also may contribute to these immune responses. Various pathogens (including Chlamydia pneumonia , Helicobacter pylori , and cytomegalovirus) have been associated with AMD in patients or in animal models. Although infection is not likely to be sufficient to cause AMD, it certainly has the potential to trigger chronic inflammation and other immune mechanisms, which in turn may initiate or drive disease processes. Autoantibodies also have been detected in the sera of patients with AMD, although it is unclear if these autoantibodies contribute to AMD pathogenesis or reflect a nonspecific immune response to retinal damage. It has been surmised that they may compromise the blood-retinal barrier.
Inflammation and drusen formation
Several studies indicate that drusen are sites of immune complex formation in AMD pathogenesis. Immunoreactivity to immunoglobulin (Ig) M, IgG, and Ig light chain have been observed in drusen. Johnson and associates demonstrated that small hard drusen (10 to 30 μm in diameter) and surrounding RPE cells exhibit immunoreactivity to IgG, whereas larger drusen (50 μm or more ) typically do not. In addition, an array of molecules involved in cellular and humoral immune response have been detected in drusen. Hageman and associates identified cells of immune origin from the choroid extending processes into drusen cores. Finally, as discussed below, molecules of the complement pathway have been identified in drusen, the Bruch membrane, and choroidal vessels. In this inflammation hypothesis of drusen formation, it is suggested that RPE cell injury is a primary event, occurring because of gene mutations, light damage, oxidative stress, lipofuscin accumulation, complement-mediated injury, or a combination thereof; injured RPE cells then recruit and activate dendritic cells from the choroid, which in turn elicit either an inflammatory- or complement-mediated response to generate drusen. Interestingly, this model does not incorporate lipid deposition as playing a role in pathogenesis.
Reticular pseudodrusen, or subretinal drusenoid deposits, also may represent changes wrought by inflammatory processes in the retina in AMD. Although these reticular deposits initially were believed to be secondary to choroidal vessel abnormalities, spectral-domain OCT reveals an undulating pattern of deposits above the RPE and extending up into the photoreceptor outer and inner segments, as shown in Figure 1 (Middle row). The pseudodrusen do not fluoresce on angiography, but are evident on red-free images and infrared imaging by scanning laser ophthalmoscope. It is possible that these changes are evidence of photoreceptor neurodegeneration, mediated by infiltrating inflammatory cells or resident microglia. They are also reminiscent of the changes seen in the CX3CR/CCl2 mouse, which initially were thought to be drusen, but actually are located in the outer retina and seem to correlate with the naturally occurring rd8 mutation in the background C57BL/6N strain.
Inflammasome
The recent discovery of the inflammasome, another component of the innate immune system, adds another aspect to the role of inflammation in AMD. The inflammasome system monitors cell stress through pattern recognition receptors, which recognize molecular conformations such as the pathogen-associated molecular patterns and damage-associated molecular patterns. Examples of the pattern recognition receptors are the Toll-like receptors (TLRs), nod-like receptors (NLRs), the C-type leptin receptors, and the virus-specific RIG-I–like receptors. Inflammasomes are multiprotein complexes that assemble in response to these signals and result in the activation of inflammatory caspases. The NLR pyrin domain-containing 3 (NLRP3) inflammasome has been shown to be activated by infection, injury, or other inflammatory disorders. Endogenous aggregates, such as uric acid crystals, amyloid polypeptides in diabetes, cholesterol crystals, and saturated fatty acids, also have been shown to induce the NRLP3 inflammasome. Tarallo and associates recently identified AluRNA (single- and double-stranded RNA) as an endogenous nucleic acid that can activate NLRP3 in the RPE. They demonstrated that AluRNA may have a role in RPE degeneration and the progression of GA, acting through the inflammatory cytokine interleukin 18 (IL-18) to activate the myeloid differentiation primary response 88 ( MYD88 ) gene. In a separate study, Doyle and associates demonstrated a different role for the inflammasome in neovascular AMD. They found that drusen (isolated from donors with AMD) activated the NLRP3 inflammasome and IL-18 expression in cultured monocytes. Using a mouse model of laser-induced CNV, they found colocalization of cleaved NRLP3 and caspase 1 with activated macrophages, and that NRLP3 protects against laser-induced CNV by activating IL-18. This protective effect of IL-18 in AMD seems to contradict not only the study by Tarallo and associates, but also previous studies implicating IL-18 and other interleukins in the induction of experimental retinal neovascularization. Although further study is required to resolve these seemingly contradictory findings, the disparity may be the result of different pathogenic mechanisms in retinal and choroidal neovascular diseases. For example, RPE dysfunction underlies the pathologic features of CNV but not retinal neovascularization. Moreover, one can speculate that the site of inflammasome activation (monocytes vs RPE) may account for at least some of the observed differences in NRLP3/IL-18 action, especially in neovascular versus nonneovascular AMD. Regardless, the available evidence supports the notion that accumulated material in the Bruch membrane (including lipids, lipofuscin components like A2E, and amyloid) can activate the NLRP3 inflammasome, leading to a chronic inflammatory response and progression to advanced forms of AMD.
Complement
Accumulating evidence strongly supports a pivotal role for complement in the pathogenesis of AMD. The complement system is a major component of innate immunity and plays well-established roles in host defense, cellular homeostasis, tissue remodeling, and tissue repair. The complement system has been implicated in a number of pathologic processes, and over the last 2 decades, several groups (reviewed by Anderson and associates ) have identified multiple complement proteins in tissues of eyes with AMD, including drusen, RPE, the Bruch membrane, basal deposits, retina, and choroidal capillaries. Additionally, the complement pathway has received increasing attention in AMD pathogenesis with the association of complement gene variants with AMD, discussed below.
The complement system (reviewed by Ricklin and associates ) consists of more than 20 protein components and 3 distinct pathways (classical, alternative, and lectin) that culminate in the activation of the central component 3 (C3) and its cleavage into C3a and C3b fragments. C3b initiates the common terminal (lytic) pathway by cleaving component 5 (C5) to C5a and C5b; component 6 then binds to C5b to catalyze the stepwise addition of component 7, component 8, and polymerized component 9 to form a rivet-like structure in the cell membrane (see Hadders and associates and Figure 2 ). This C5b-9 membrane attack complex (MAC) creates pores through the lipid bilayer ( Figure 2 ) that facilitates cellular lysis and clearance.
C3 contains an internal thioester bond that is unstable in aqueous environments and spontaneously hydrolyzes into C3a and C3b. Approximately a dozen proteins, including complement factor H (CFH), membrane cofactor protein, vitronectin, and CD59, act as regulators of the complement cascade to protect self cells from intrinsic complement activation. Negative regulators are particularly important with the alternative complement cascade, which uses the spontaneous lysis of C3 to maintain a continuous low-level state of activation to ensure constant surveillance. CFH is a critical regulator of the alternative pathway and prevents MAC formation on cells belonging to self via 2 mechanisms: (1) accelerating the decay of the C3 convertase (C3bBb) by removing Bb, or (2) acting as a cofactor for complement factor I (CFI), which degrades C3b. Since the discovery of a specific CFH gene variant that is strongly associated with AMD risk, the alternative pathway has garnered particular attention in the pathophysiology factors of AMD.
Although systemically circulating complement proteins are usually synthesized in hepatocytes, complement production also may occur in extrahepatic tissues, including the neural retina, RPE, and choroid. Activation of complement likely results from age-related accumulation of endogenous materials. Indeed, in vitro models of drusen formation have shown that the alternative complement pathway may be activated by the accumulation of lipofuscin components and apolipoproteins. Aβ also colocalizes histologically with sites of complement activation in AMD specimens. C5b-9 has been correlated with abnormal RPE cells, suggesting that complement-mediated cell lysis may accelerate RPE dysfunction and death in AMD.
The complement system also seems to be a critical regulator of CNV. This in part may be the result of the association of complement with the Bruch membrane, which normally acts as a barrier to CNV (see Figure 2 ). C5b-9 has been detected in the Bruch membrane of AMD donors, suggesting that complement may contribute to the breakdown of the Bruch membrane that occurs with CNV. Some complement components also display proangiogenic properties. The C5a protein, cleaved at the beginning of the terminal pathway, has been shown to promote neovascularization in a murine model of laser-induced CNV and to promote endothelial cell migration, proliferation, and vessel formation in vitro. Additionally, C5a has been shown to promote VEGF secretion in vitro. In mice deficient in either monocyte chemoattractant protein-1 or its cognate C-C chemokine receptor-2, impaired macrophage recruitment may allow C5a accumulation. Studies using knockout mice have suggested that activation of the alternative complement cascade is central to laser-induced CNV in mice. In mouse models, CNV is reduced using a soluble form of CD59 (a natural membrane-bound inhibitor of MAC assembly) and a recombinant form of CFH, which suggests that targeted inhibition of complement is a feasible therapeutic strategy for AMD (see Supplemental Material ). Interestingly, however, deficiency of the central component C3 (as well as deficiency or inhibition of the C5a axis) increased neovascularization in a murine model of retinopathy of prematurity and in an in vivo angiogenesis assay, suggesting that complement has an inhibitory role in neovascularization. Again, this seemingly contradictory finding may be the result of different pathogenic mechanisms in retinal and choroidal neovascular diseases. Nonetheless, this highlights the multifaceted roles of the complement system in normal physiology and pathogenesis.
Although complement clearly plays a role in the pathogenesis of AMD, it is not clear whether the cascade is underactive or overactive, which components are key, or how one might best intervene with drug therapy. Some insight may be gained from observations in patients with membranoproliferative glomerulonephritis type II (MPGN II), a rare disease in which abnormal electron-dense material is deposited in the glomerular basement membrane of the kidney and the Bruch membrane. The disease occurs because of chronic activation of the alternative complement pathway. In most patients, this occurs because an autoantibody called nephritic factor keeps the C3 convertase (C3bBb) constitutively active, resulting in low serum C3 levels and C3 deposits in the kidney and eye. Mutations in the CFH gene have been identified in some cases, and plasma exchange with normal CFH may be an effective treatment in this subset of MPGN II patients. Renal failure often leads to renal transplantation, but the disease recurs in nearly all cases. In patients with MPGN II, drusen develop at an early age, are often visible in the second decade, and are characterized by whitish-yellow deposits beneath the RPE. Asymptomatic in the early stages of disease, patients can demonstrate abnormalities of dark adaptation similar to AMD, with vision problems occurring in approximately 10% of patients. The retinopathy is slowly progressive and often accompanied by atrophy, although CNV also can occur. The similarities between MPGN II and AMD suggest that the latter may arise from a chronically overactive complement system, at least at the level of the outer retina.
Angiogenesis
Although the biologic process of angiogenesis may not underlie the earliest stages of disease—instead representing a programmed response to injury and inflammation in AMD—angiogenesis is an important process in advanced AMD. Neovascular AMD is characterized by vascular growth and leakage, occurring in the sub-RPE, subretinal space, and neural retina. In recent decades, major advances in vascular biology have revolutionized the therapeutic strategy for neovascular AMD. Angiogenesis, which is defined as the growth of new vessels from established vascular beds, is an important underlying mechanism of CNV; it involves endothelial cell proliferation and migration along the ECM, endothelial tube formation, and vessel maturation. ECM integrity is pivotal in AMD pathogenesis; it is believed that CNV requires a disruption in the Bruch membrane (see Figure 2 ), which can result from exogenous factors (such as mechanical disruption) or by endogenous degradation (by excessive proteolysis or immune attack, such as complement activation). Discontinuity in the Bruch membrane may allow endothelial cells to migrate into the sub-RPE space, forming immature vessels that are leaky and tortuous, and may extend into the subretinal space. VEGF, a potent endothelial cell mitogen and vascular permeability factor, is a prominent angiogenic signal that plays a pivotal role in the pathogenesis of neovascular AMD. VEGF is a secreted endothelial cell mitogen that is involved in migration and proliferation of endothelial cells, and is a key factor in the abnormal permeability of CNV. Furthermore, VEGF is known to increase levels of MMPs, which not only contribute to ECM breakdown, but also may increase VEGF expression and secretion from RPE cells further via a feedback mechanism. Current successful therapies for neovascular AMD are based on VEGF inhibition.
Cell Death Pathways
Although the different forms of advanced AMD have distinct pathologic mechanisms, they converge on cellular pathways that lead to photoreceptor death (see Figure 3 ). Photoreceptor death, which is the ultimate cause of vision loss in AMD, also can occur through multiple distinct pathways. Apoptosis (Greek for “falling off”) is the best-known mechanism of controlled cell death and is regulated by the caspase family of enzymes. Cells undergoing apoptosis are rounded and shrunken; the nuclear DNA becomes condensed and fragmented, and white blood cells engulf the apoptotic cells before they rupture and cause inflammation. Although many studies have focused on apoptosis as the sole mechanism of photoreceptor death, necrosis is emerging as an important cell death pathway in retinal degenerative diseases. In contrast to apoptosis, necrosis (Greek for “dead”) is characterized by cell swelling and rupture. Previously believed to be an uncontrolled mechanism of cell death, necrosis is now known to be regulated in part by the receptor interacting protein (RIP) kinase family of enzymes. We recently demonstrated that controlled necrosis (termed necroptosis ) is a significant method of photoreceptor death in the retina and compensates for a reduction in apoptosis when caspases are inhibited. Moreover, RIP-mediated necrosis is involved in cone (but not rod) cell death in a mouse model of retinal degeneration. Thus, it seems that both apoptosis and necroptosis lead to photoreceptor death, and targeting both pathways may be an effective neuroprotective strategy for retinal disorders.
Pathogenesis: An Evolving Story
Our understanding of AMD and its pathogenesis has changed over decades of observation. Although there has been progress, there are still missing pieces in the puzzle, and any attempt at a unified model will meet with disagreement. Nevertheless, formulating a model is a worthwhile exercise and a prerequisite to developing biologically based therapies. However, before developing a model based on current knowledge, it is useful to review the pathogenesis of AMD from a historical perspective.
Senile macular degeneration first was described as a clinical disorder by Haab in 1885 (reviewed by Gass ), but for decades, clinicians did not make the connection between macular drusen and disciform scars, which represent the end stage of exudative AMD ( Figure 1 , Bottom right). J. Donald Gass, the keen observer here as he was for so many retinal entities, described the progression of macular drusen to serous RPE, retinal detachments, and neovascularization in the subretinal and sub-RPE space, as well as describing GA. Many of Gass’ early observations remain clinically relevant. Patients with extramacular or peripheral drusen, or both, sparing the macula rarely lose vision, and macular drusen often disappear with the onset of serous detachments or neovascularization. Gass also noted that the disorder was more common in families, supporting a genetic contribution, and he believed that GA and neovascularization were different manifestations of the same disease. Although some have questioned whether the different phenotypes represent different diseases, Gass’ conclusion—that all are related forms of AMD—is probably correct. Figure 3 illustrates this concept and emphasizes the interconnections of AMD stages.
Epidemiologic studies, including the Framingham Eye study, First National Health and Nutrition Examination Survey, and the Eye Disease Case-Control Study, sought to understand the pathogenesis of AMD by evaluating risk factors. Advanced age is the greatest risk factor for AMD, and cigarette smoking remains the most consistent preventable risk factor across all studies. Associations with cardiovascular risk factors, although varying individually, are consistent as a group; for instance, hypertension was found to be associated in some studies, whereas elevated cholesterol was associated in others. Although an association has been postulated between ultraviolet light exposure and AMD, the Eye Disease Case-Control Study found no association. This was in agreement with previous studies by West and associates, who examined a cohort of Chesapeake Bay watermen and found no association between ultraviolet B or ultraviolet A exposure and AMD. However, Taylor and associates did find an association between visible light exposure and advanced AMD (GA or neovascular AMD). Hyperopia was identified as a risk factor for CNV in a 1993 study conducted at Massachusetts Eye and Ear Infirmary, a finding that was corroborated by several large studies–notably AREDS, the Rotterdam Study, and the Beijing Eye Study. It has been suggested that hyperopic shortening may lead to abnormal choroidal perfusion and increased choroidal resistance, thus predisposing eyes with shorter axial lengths to AMD. Higher serum levels of carotenoids, large horizontal cup-to-disc ratios, and postmenopausal estrogen use in women were associated with a reduced risk of neovascular AMD. Risk factors that have been suggested variably but are no longer a subject of focus include left ventricular hypertrophy, decreased hand grip strength, short height, decreased vital capacity, and history of lung infection, as well as parity greater than zero for neovascular AMD.
Clinical observations and epidemiologic data stimulated the development of mechanistic models of AMD pathogenesis. Further clinical pathologic correlations and biochemical investigations contributed to these ideas. The changes observed in early AMD are now known to involve many physiologic processes that may become pathologic over time and may be exacerbated by genetic or environmental factors.
Age and Lipid Deposition
Age is the greatest risk factor for AMD, and even with normal aging, the Bruch membrane undergoes neutral lipid deposition, as demonstrated by Pauleikhoff and associates in 1990. The notion of lipid accumulation as an early harbinger of AMD remains a current concept and invokes comparisons with atherosclerosis, another age-related disease with both hemodynamic and lipid-based hypotheses. However, the lipid-rich lesions (such as BlinD and drusen) are distinctive to AMD and do not necessarily reflect an aging process. Thus, understanding what is different about lipid accumulation in AMD should provide insight into the pathogenesis of early AMD.
On the basis of its binding with oil red-O, neutral lipid was identified in drusen in the 1960s, and emerging evidence suggests that lipid accumulation has an underlying role in the pathogenesis of AMD. Studying the hydrodynamics of the Bruch membrane using donor tissues, Starita and associates demonstrated that hydraulic conductivity declines with age, and that impaired transport resulting from lipid deposition may contribute to AMD. More recently, it has become apparent that deposits in the Bruch membrane likely arise from multiple sources, including outer segments debris, systemic circulation, and endogenous synthesis. Having demonstrated through accumulated evidence that RPE is the likely source for lipoprotein accumulation in both aging and in AMD, Curcio and associates formulated a compelling model for AMD pathogenesis. According to their model, the Bruch membrane functions much like the endothelium in atherosclerosis, and parallel processes in AMD and atherosclerosis can be traced to lipid deposition. Specifically, lipoproteins such as apolipoprotein B, which serve the biological function of delivering cholesterol to tissues, become retained in the subendothelium of arteries. Oxidative and nonoxidative modifications subsequently trigger pathologic adaptive responses. This model of atherosclerosis, known as the response-to-retention hypothesis, provides a feasible explanation for comparable processes in AMD: apolipoprotein B and other lipids (likely produced by the RPE) accumulate in the Bruch membrane and sub-RPE space, and pathologic responses to these retained particles lead to AMD. Thus, whereas atherosclerosis is the consequence for relying on apolipoprotein B for cholesterol and lipid transport, AMD is the consequence for relying on similar systems to transport fat-soluble nutrients (such as retinol) in the retina.
Examining age-related changes in human eyes using quick-freeze or deep-etch techniques, Ruberti and associates observed the accumulation of esterified cholesterol and unesterified cholesterol in the Bruch membrane. They showed that in early adulthood, esterified cholesterol- and unesterified cholesterol-containing particles form a thin sublayer; by 60 years of age, a confluent, hydrophobic barrier forms, with the esterified cholesterol and unesterified cholesterol concentrated in macular regions. Because the observed lipid wall coincides with the location of BlinD (see Figure 2 ), which are characteristic of early AMD, it is likely that the lipid wall precedes the formation of BlinD and drusen. Further bolstering this model, Curcio and associates showed that drusen are composed of at least 40% lipid. These observations support the hypothesis that AMD, like atherosclerosis, has a pathologic component that involves lipid deposition.
Photoreceptor distribution also changes with age. The macula is characterized by a cone-dominated fovea surrounded by a rod-dominated parafovea. Cones are stable in adulthood, but rods decrease by 30%, with even greater loss overlying sub-RPE deposits. Interestingly, as the rods die, the surviving rod inner segments expand to fill the vacated space, suggesting that the rods and cones actively regulate their inner segment space allocation. RPE cell morphologic features also show topographical and age-related differences. Early in life, the RPE cells of the macular area are broader and lower, becoming narrower and higher toward the equator and periphery. In older eyes, this gradient is reversed: cells in the macular area become narrower and higher, whereas cells in the periphery become broader and lower with little change in the equatorial zone.
Vascular Resistance
Based on the similarities between AMD and atherosclerosis, Friedman postulated that hemodynamic changes and increased choroidal resistance underlie the pathogenesis of AMD. He traced the hypothesis back to Friedenwald’s observation that scleral rigidity increased with age, and to Verhoeff and Grossman’s observation that AMD seemed to be associated with drusen and impaired choroidal flow. Friedman showed that increased scleral rigidity was associated with AMD compared with age-matched controls and attributed this increase in scleral rigidity to lipid deposition—similar to atherosclerosis. He had observed that drusen appeared in the posterior pole and adjacent to vortex veins anteriorly, where the venous pressure is greatest, suggesting that their formation relates in part to a venous abnormality. Friedman postulated that progressive infiltration by lipids in the sclera and other ocular tissues leads to increased resistance in the choroidal vessels, thereby diminishing the ability of the choriocapillaris to clear lipoproteins from the RPE and Bruch membrane. Hyperopia, a risk factor previously identified, would contribute to increased resistance in choroidal vessels, and thus supports the hemodynamic model of AMD. Choriocapillaris density also has been shown to be increased significantly in both neovascular and nonneovascular AMD compared with controls, suggesting increased resistance and impaired flow. As a consequence of these changes, metabolic exchange across the RPE may be compromised, and lipoproteins from photoreceptors and other sources could accumulate in the Bruch membrane as well as the outer retina to form drusen. In proximity to the RPE, these alterations could lead to atrophy; additionally, by altering the Bruch membrane with subsequent calcification and fracture, the accumulation could lead to neovascularization.
Lipofuscin Accumulation
Light-sensitive photoreceptor outer segments are renewed constantly, and the RPE functions to phagocytose photoreceptor outer segment discs for lysosomal degradation. In the 1970s, Hogan surmised that age-related changes in the Bruch membrane and the RPE result from the accumulation of by-products of this phagocytic process. Lipofuscin, a granular pigment that results from lysosomal digestion, has been shown to accumulate in the RPE with age. Lipofuscin accumulation in the RPE results in increased fundus autofluorescence, which has been suggested to be a prelude to GA. Lipofuscin is not defined completely, but several fluorophores have been isolated. One important fluorophore is A2E, a pyridinium bis-retinoid composed of 2 vitamin A aldehyde molecules and 1 ethanolamine molecule. A2E accounts for the fluorescent properties of lipofuscin and forms the basis of clinical fundus autofluorescence imaging. A2E has been shown to inhibit lysosomal degradative function and cholesterol metabolism in the RPE. A2E is capable of acting as a photosensitizer and has been shown to mediate blue-light-induced apoptosis. Furthermore, A2E has been demonstrated to induce the complement system of innate immunity, which has an emerging role in AMD pathogenesis (discussed below). Via these mechanisms, A2E may have toxic effects on the RPE, leading to cell death and atrophy. In murine models of macular degeneration and other retinal degenerative disorders, lipofuscin accumulation often is associated with neural degeneration (reviewed in Ramkumar and associates ). Although lipofuscin accumulation is central to the pathologic process, determining whether this is cause or effect is critical to understanding the initiation of AMD.
Amyloid Accumulation
Several studies have shown that similar processes cause drusen formation in the eye and senile plaque formation in the brain, invoking comparisons between AMD and another age-related condition: Alzheimer disease. Both diseases exhibit the accumulation of extracellular deposits such as apolipoprotein E (ApoE) and amyloid-beta (Aβ). ApoE functions in normal lipid catabolism and transport and is a major lipoprotein component of drusen. Variants of the APOE gene have been implicated in Alzheimer disease as well as AMD, although the risk associations are in opposite directions. The E4 allele is a risk factor for Alzheimer disease, but seems to be protective for AMD, whereas the E2 allele is a risk factor for AMD. Aβ, a peptide that forms a large proportion of senile plaques, has been identified as a significant component in drusen and has been implicated in inflammatory events (discussed below) that contribute to RPE dysfunction in AMD. Subsequent studies have shown that Aβ-containing drusen are specific to the retinas of AMD patients and seem to correlate with advanced stages of AMD. There may be a genetic link between AMD and Alzheimer disease in the amyloid pathway, because a gene variant of cystatin C (believed to be involved in amyloid deposition) has been linked to increased susceptibility to both diseases. Using an antibody to target Aβ peptides in the APOE4-HFC mouse model of AMD, Marcu and associates demonstrated reduced levels of both Aβ and activated complement components in sub-RPE deposits and structural preservation of the RPE. According to in vitro studies, AMD-related Aβ is likely of RPE origin and is proangiogenic, inducing VEGF expression while decreasing the expression of pigment epithelium-derived factor (PEDF), an endogenous antiangiogenic protein. Interestingly, Aβ recently was demonstrated to accumulate in photoreceptor outer segments as an age-related change in normal human retinas, suggesting that it may be associated with visual function at the level of the neural retina. Although AMD and Alzheimer disease both exhibit apolipoprotein and amyloid deposition, several differences remain, including the disparity of genetic risk factors. Moreover, just as the amyloid hypothesis for Alzheimer disease has been questioned, it remains unclear whether Aβ plays a causative role in AMD.
Inflammation and Immune Response
In addition to age-related changes such as hemodynamic alterations and lipid accumulation, inflammation is recognized widely as another important player in the pathogenesis of both atherosclerosis and AMD. Genetic variation in the complement pathway (discussed below) and environmental factors (like cigarette smoking) may predispose individuals to the disease through inflammatory mechanisms. Moreover, inflammatory responses can upregulate VEGF and other proangiogenic factors that are essential for CNV, linking inflammation with the progression of AMD to advanced stages.
Long before the link between AMD and complement gene variants became widely recognized, Hageman and associates were vigorous proponents of a role for inflammation in the pathogenesis of AMD, based on observations of human specimens. Hageman and associates postulated that inflammatory or immune-mediated processes (including the recruitment and activation of dendritic cells) were central to the development of drusen and subsequent events. This notion is supported by clinicopathologic studies: multinucleated giant cells, mononuclear phagocyte system (MPS) cells, various immunocompetent cells, and increased HLA-DR immunoreactivity in retinal microglia have been described in histopathologic specimens from AMD patients. Age-related changes in retinal microglial activation, which have been linked to lipofuscin accumulation, may contribute to immune dysfunction in AMD pathogenesis. Infectious agents also may contribute to these immune responses. Various pathogens (including Chlamydia pneumonia , Helicobacter pylori , and cytomegalovirus) have been associated with AMD in patients or in animal models. Although infection is not likely to be sufficient to cause AMD, it certainly has the potential to trigger chronic inflammation and other immune mechanisms, which in turn may initiate or drive disease processes. Autoantibodies also have been detected in the sera of patients with AMD, although it is unclear if these autoantibodies contribute to AMD pathogenesis or reflect a nonspecific immune response to retinal damage. It has been surmised that they may compromise the blood-retinal barrier.
Inflammation and drusen formation
Several studies indicate that drusen are sites of immune complex formation in AMD pathogenesis. Immunoreactivity to immunoglobulin (Ig) M, IgG, and Ig light chain have been observed in drusen. Johnson and associates demonstrated that small hard drusen (10 to 30 μm in diameter) and surrounding RPE cells exhibit immunoreactivity to IgG, whereas larger drusen (50 μm or more ) typically do not. In addition, an array of molecules involved in cellular and humoral immune response have been detected in drusen. Hageman and associates identified cells of immune origin from the choroid extending processes into drusen cores. Finally, as discussed below, molecules of the complement pathway have been identified in drusen, the Bruch membrane, and choroidal vessels. In this inflammation hypothesis of drusen formation, it is suggested that RPE cell injury is a primary event, occurring because of gene mutations, light damage, oxidative stress, lipofuscin accumulation, complement-mediated injury, or a combination thereof; injured RPE cells then recruit and activate dendritic cells from the choroid, which in turn elicit either an inflammatory- or complement-mediated response to generate drusen. Interestingly, this model does not incorporate lipid deposition as playing a role in pathogenesis.
Reticular pseudodrusen, or subretinal drusenoid deposits, also may represent changes wrought by inflammatory processes in the retina in AMD. Although these reticular deposits initially were believed to be secondary to choroidal vessel abnormalities, spectral-domain OCT reveals an undulating pattern of deposits above the RPE and extending up into the photoreceptor outer and inner segments, as shown in Figure 1 (Middle row). The pseudodrusen do not fluoresce on angiography, but are evident on red-free images and infrared imaging by scanning laser ophthalmoscope. It is possible that these changes are evidence of photoreceptor neurodegeneration, mediated by infiltrating inflammatory cells or resident microglia. They are also reminiscent of the changes seen in the CX3CR/CCl2 mouse, which initially were thought to be drusen, but actually are located in the outer retina and seem to correlate with the naturally occurring rd8 mutation in the background C57BL/6N strain.
Inflammasome
The recent discovery of the inflammasome, another component of the innate immune system, adds another aspect to the role of inflammation in AMD. The inflammasome system monitors cell stress through pattern recognition receptors, which recognize molecular conformations such as the pathogen-associated molecular patterns and damage-associated molecular patterns. Examples of the pattern recognition receptors are the Toll-like receptors (TLRs), nod-like receptors (NLRs), the C-type leptin receptors, and the virus-specific RIG-I–like receptors. Inflammasomes are multiprotein complexes that assemble in response to these signals and result in the activation of inflammatory caspases. The NLR pyrin domain-containing 3 (NLRP3) inflammasome has been shown to be activated by infection, injury, or other inflammatory disorders. Endogenous aggregates, such as uric acid crystals, amyloid polypeptides in diabetes, cholesterol crystals, and saturated fatty acids, also have been shown to induce the NRLP3 inflammasome. Tarallo and associates recently identified AluRNA (single- and double-stranded RNA) as an endogenous nucleic acid that can activate NLRP3 in the RPE. They demonstrated that AluRNA may have a role in RPE degeneration and the progression of GA, acting through the inflammatory cytokine interleukin 18 (IL-18) to activate the myeloid differentiation primary response 88 ( MYD88 ) gene. In a separate study, Doyle and associates demonstrated a different role for the inflammasome in neovascular AMD. They found that drusen (isolated from donors with AMD) activated the NLRP3 inflammasome and IL-18 expression in cultured monocytes. Using a mouse model of laser-induced CNV, they found colocalization of cleaved NRLP3 and caspase 1 with activated macrophages, and that NRLP3 protects against laser-induced CNV by activating IL-18. This protective effect of IL-18 in AMD seems to contradict not only the study by Tarallo and associates, but also previous studies implicating IL-18 and other interleukins in the induction of experimental retinal neovascularization. Although further study is required to resolve these seemingly contradictory findings, the disparity may be the result of different pathogenic mechanisms in retinal and choroidal neovascular diseases. For example, RPE dysfunction underlies the pathologic features of CNV but not retinal neovascularization. Moreover, one can speculate that the site of inflammasome activation (monocytes vs RPE) may account for at least some of the observed differences in NRLP3/IL-18 action, especially in neovascular versus nonneovascular AMD. Regardless, the available evidence supports the notion that accumulated material in the Bruch membrane (including lipids, lipofuscin components like A2E, and amyloid) can activate the NLRP3 inflammasome, leading to a chronic inflammatory response and progression to advanced forms of AMD.
Complement
Accumulating evidence strongly supports a pivotal role for complement in the pathogenesis of AMD. The complement system is a major component of innate immunity and plays well-established roles in host defense, cellular homeostasis, tissue remodeling, and tissue repair. The complement system has been implicated in a number of pathologic processes, and over the last 2 decades, several groups (reviewed by Anderson and associates ) have identified multiple complement proteins in tissues of eyes with AMD, including drusen, RPE, the Bruch membrane, basal deposits, retina, and choroidal capillaries. Additionally, the complement pathway has received increasing attention in AMD pathogenesis with the association of complement gene variants with AMD, discussed below.
The complement system (reviewed by Ricklin and associates ) consists of more than 20 protein components and 3 distinct pathways (classical, alternative, and lectin) that culminate in the activation of the central component 3 (C3) and its cleavage into C3a and C3b fragments. C3b initiates the common terminal (lytic) pathway by cleaving component 5 (C5) to C5a and C5b; component 6 then binds to C5b to catalyze the stepwise addition of component 7, component 8, and polymerized component 9 to form a rivet-like structure in the cell membrane (see Hadders and associates and Figure 2 ). This C5b-9 membrane attack complex (MAC) creates pores through the lipid bilayer ( Figure 2 ) that facilitates cellular lysis and clearance.
C3 contains an internal thioester bond that is unstable in aqueous environments and spontaneously hydrolyzes into C3a and C3b. Approximately a dozen proteins, including complement factor H (CFH), membrane cofactor protein, vitronectin, and CD59, act as regulators of the complement cascade to protect self cells from intrinsic complement activation. Negative regulators are particularly important with the alternative complement cascade, which uses the spontaneous lysis of C3 to maintain a continuous low-level state of activation to ensure constant surveillance. CFH is a critical regulator of the alternative pathway and prevents MAC formation on cells belonging to self via 2 mechanisms: (1) accelerating the decay of the C3 convertase (C3bBb) by removing Bb, or (2) acting as a cofactor for complement factor I (CFI), which degrades C3b. Since the discovery of a specific CFH gene variant that is strongly associated with AMD risk, the alternative pathway has garnered particular attention in the pathophysiology factors of AMD.
Although systemically circulating complement proteins are usually synthesized in hepatocytes, complement production also may occur in extrahepatic tissues, including the neural retina, RPE, and choroid. Activation of complement likely results from age-related accumulation of endogenous materials. Indeed, in vitro models of drusen formation have shown that the alternative complement pathway may be activated by the accumulation of lipofuscin components and apolipoproteins. Aβ also colocalizes histologically with sites of complement activation in AMD specimens. C5b-9 has been correlated with abnormal RPE cells, suggesting that complement-mediated cell lysis may accelerate RPE dysfunction and death in AMD.
The complement system also seems to be a critical regulator of CNV. This in part may be the result of the association of complement with the Bruch membrane, which normally acts as a barrier to CNV (see Figure 2 ). C5b-9 has been detected in the Bruch membrane of AMD donors, suggesting that complement may contribute to the breakdown of the Bruch membrane that occurs with CNV. Some complement components also display proangiogenic properties. The C5a protein, cleaved at the beginning of the terminal pathway, has been shown to promote neovascularization in a murine model of laser-induced CNV and to promote endothelial cell migration, proliferation, and vessel formation in vitro. Additionally, C5a has been shown to promote VEGF secretion in vitro. In mice deficient in either monocyte chemoattractant protein-1 or its cognate C-C chemokine receptor-2, impaired macrophage recruitment may allow C5a accumulation. Studies using knockout mice have suggested that activation of the alternative complement cascade is central to laser-induced CNV in mice. In mouse models, CNV is reduced using a soluble form of CD59 (a natural membrane-bound inhibitor of MAC assembly) and a recombinant form of CFH, which suggests that targeted inhibition of complement is a feasible therapeutic strategy for AMD (see Supplemental Material ). Interestingly, however, deficiency of the central component C3 (as well as deficiency or inhibition of the C5a axis) increased neovascularization in a murine model of retinopathy of prematurity and in an in vivo angiogenesis assay, suggesting that complement has an inhibitory role in neovascularization. Again, this seemingly contradictory finding may be the result of different pathogenic mechanisms in retinal and choroidal neovascular diseases. Nonetheless, this highlights the multifaceted roles of the complement system in normal physiology and pathogenesis.
Although complement clearly plays a role in the pathogenesis of AMD, it is not clear whether the cascade is underactive or overactive, which components are key, or how one might best intervene with drug therapy. Some insight may be gained from observations in patients with membranoproliferative glomerulonephritis type II (MPGN II), a rare disease in which abnormal electron-dense material is deposited in the glomerular basement membrane of the kidney and the Bruch membrane. The disease occurs because of chronic activation of the alternative complement pathway. In most patients, this occurs because an autoantibody called nephritic factor keeps the C3 convertase (C3bBb) constitutively active, resulting in low serum C3 levels and C3 deposits in the kidney and eye. Mutations in the CFH gene have been identified in some cases, and plasma exchange with normal CFH may be an effective treatment in this subset of MPGN II patients. Renal failure often leads to renal transplantation, but the disease recurs in nearly all cases. In patients with MPGN II, drusen develop at an early age, are often visible in the second decade, and are characterized by whitish-yellow deposits beneath the RPE. Asymptomatic in the early stages of disease, patients can demonstrate abnormalities of dark adaptation similar to AMD, with vision problems occurring in approximately 10% of patients. The retinopathy is slowly progressive and often accompanied by atrophy, although CNV also can occur. The similarities between MPGN II and AMD suggest that the latter may arise from a chronically overactive complement system, at least at the level of the outer retina.
Angiogenesis
Although the biologic process of angiogenesis may not underlie the earliest stages of disease—instead representing a programmed response to injury and inflammation in AMD—angiogenesis is an important process in advanced AMD. Neovascular AMD is characterized by vascular growth and leakage, occurring in the sub-RPE, subretinal space, and neural retina. In recent decades, major advances in vascular biology have revolutionized the therapeutic strategy for neovascular AMD. Angiogenesis, which is defined as the growth of new vessels from established vascular beds, is an important underlying mechanism of CNV; it involves endothelial cell proliferation and migration along the ECM, endothelial tube formation, and vessel maturation. ECM integrity is pivotal in AMD pathogenesis; it is believed that CNV requires a disruption in the Bruch membrane (see Figure 2 ), which can result from exogenous factors (such as mechanical disruption) or by endogenous degradation (by excessive proteolysis or immune attack, such as complement activation). Discontinuity in the Bruch membrane may allow endothelial cells to migrate into the sub-RPE space, forming immature vessels that are leaky and tortuous, and may extend into the subretinal space. VEGF, a potent endothelial cell mitogen and vascular permeability factor, is a prominent angiogenic signal that plays a pivotal role in the pathogenesis of neovascular AMD. VEGF is a secreted endothelial cell mitogen that is involved in migration and proliferation of endothelial cells, and is a key factor in the abnormal permeability of CNV. Furthermore, VEGF is known to increase levels of MMPs, which not only contribute to ECM breakdown, but also may increase VEGF expression and secretion from RPE cells further via a feedback mechanism. Current successful therapies for neovascular AMD are based on VEGF inhibition.
Cell Death Pathways
Although the different forms of advanced AMD have distinct pathologic mechanisms, they converge on cellular pathways that lead to photoreceptor death (see Figure 3 ). Photoreceptor death, which is the ultimate cause of vision loss in AMD, also can occur through multiple distinct pathways. Apoptosis (Greek for “falling off”) is the best-known mechanism of controlled cell death and is regulated by the caspase family of enzymes. Cells undergoing apoptosis are rounded and shrunken; the nuclear DNA becomes condensed and fragmented, and white blood cells engulf the apoptotic cells before they rupture and cause inflammation. Although many studies have focused on apoptosis as the sole mechanism of photoreceptor death, necrosis is emerging as an important cell death pathway in retinal degenerative diseases. In contrast to apoptosis, necrosis (Greek for “dead”) is characterized by cell swelling and rupture. Previously believed to be an uncontrolled mechanism of cell death, necrosis is now known to be regulated in part by the receptor interacting protein (RIP) kinase family of enzymes. We recently demonstrated that controlled necrosis (termed necroptosis ) is a significant method of photoreceptor death in the retina and compensates for a reduction in apoptosis when caspases are inhibited. Moreover, RIP-mediated necrosis is involved in cone (but not rod) cell death in a mouse model of retinal degeneration. Thus, it seems that both apoptosis and necroptosis lead to photoreceptor death, and targeting both pathways may be an effective neuroprotective strategy for retinal disorders.
Genetics of Age-Related Macular Degeneration
Clinical observations, epidemiologic data, clinicopathologic correlations, and biochemical studies have provided a large body of evidence on which to base models of AMD pathogenesis. The recent revolution in genetic and genomic studies has contributed significantly to our understanding of AMD pathogenesis—in some cases confirming factors identified by other means, and in other cases, identifying new pathways.
Genetic Risk Factors for Age-Related Macular Degeneration Susceptibility
The genetic predisposition for AMD has been recognized for some time. Gass noted that the disorder tended to cluster in families, and Francois noted the concordance of senile macular degeneration in monozygotic twins, observing both autosomal dominant and recessive forms with variable penetration. These observations since have been confirmed by a number of population-based studies showing familial aggregation, twin studies demonstrating a higher concordance of AMD in monozygotic than dizygotic twins, and the more recent discovery of gene loci that modify AMD susceptibility (reviewed by DeAngelis and associates).
AMD is a challenge to study from a genetic standpoint for several reasons. The disease appears relatively late in life, so the parents and older siblings of the index patient no longer may be living, and descendants of the index patient may not yet show any signs of the disease. AMD has a high prevalence, especially in the older white population, affecting more than 25% of people by the eighth decade of life. It is becoming clear that many genes contribute to AMD and may involve complex interactions with other genetic or environmental factors. The study of AMD is complicated further by the fact that it is a heterogeneous disease with both neovascular and nonneovascular forms and other possible important subtypes within these categories. Furthermore, the phenotypic characterization of AMD progression still needs refinement, complicating efforts to correlate genotype and phenotype.
Several approaches have been used to identify genetic loci that contribute to AMD risk. Genome-wide association studies (which use both unrelated case controls and family-based cohorts) and linkage studies (which use families only) have identified a number of loci. In some cases, genes involved in Mendelian-inherited macular degenerations have also shown association with AMD. In reviewing the genetic literature, it is apparent that genetic findings seem to differ between investigators and between different study populations. This may be explained partly by the fact that single nucleotide polymorphisms (SNPs) may vary in the minor allele frequency among different ethnic populations. Moreover, SNPs serve as markers for particular gene regions and usually do not represent the actual disease-contributing mutations. Rather association often result from linkage disequilibrium or nonrandom cosegregation with the causal mutations. Thus far, our genetic studies have identified variants in genes associated with AMD susceptibility, and we now need to direct our focus to the specific mutations and variants that actually cause disease. Using a systems-based analysis of the genes implicated in AMD, we can gain a better understanding of the disease and may explain its heterogeneity.
Two Major Loci
In the past decade, extensive genetic evidence has centered around 2 loci: 1q32 and 10q26 ( Figure 4 ). The 1q32 locus contains several functionally related genes important in inflammation and immune function, known collectively as the regulators of complement activation (RCA) gene cluster. As such, these genes encode important regulators of the complement system and may play roles in other pathways as well. Confirming the importance of complement in AMD pathogenesis, polymorphisms in all RCA genes have been associated with AMD risk.
CFH (1q32)
As previously discussed, CFH is a critical regulator of the alternative complement pathway. In 2005, several groups found strong associations between the Y402H polymorphism in the CFH gene and AMD susceptibility, and this association has since been replicated in various populations and confirmed by multiple meta-analyses. Dysregulation of CFH may contribute to many pathogenic features of AMD. This is evidenced by chimeric mice expressing the human 402H variant, which causes features similar to the intermediate stages of AMD to develop, such as basal deposits, increased lipofuscin, and subretinal drusenoid deposits. The population attributable risk of the CFH Y402H variant has been reported to be 25% to 50% or higher, indicating a substantial contribution of this locus to AMD susceptibility. Although this variant is less common in Asian populations, pooled analyses also have supported an association between the Y402H allele and AMD in these populations. Direct sequencing of the CFH gene has identified a rare but highly penetrant mutation (H5) that confers a high risk of AMD and increased severity with early onset of disease, and CFH polymorphisms also have been associated with the PCV phenotype.
CFHR1–5 (1q32–1q31.3)
Five genes encoding CFH-related (CFHR) proteins within the RCA gene cluster are believed to have resulted from gene duplication events. The CFHRs lack the N-terminal complement regulatory region of CFH, and thus may not exhibit the same degree of cofactor and decay-accelerating activity. However, CFH and CFHRs have related C-terminal surface binding regions, and sequence variations within these regions may confer different surface-binding affinities. CFHR3, CFHR4, and CFHR5 can each bind C3b and are believed to augment CFH-mediated C3b degradation. CFHR1 has been shown to block C5 convertase activity, which would interfere with C5b deposition and MAC formation on surfaces. Conversely, CFHR1 also may inhibit CFH activity by competing with surface binding. The effects of CFHR polymorphisms on overall AMD risk thus may depend on complex interactions among the CFHR and CFH genes. This is apparent in the contrasting phenotypes associated with CFHR gene deletions, also known as copy number variations. For example, a common deletion that spans the adjacent CFHR1 and CFHR3 genes has been shown to decrease AMD risk, and this deletion may interact with protective CFH haplotypes. In contrast, a rare deletion of CFHR1 and CFHR4 has been associated with bilateral GA. Obviously, it will require further analysis to explain the complex interactions implied by these observations.
F13B (1q31–q32.1)
Part of the RCA gene cluster on chromosome 1q32 ( Figure 4 ), F13B encodes the noncatalytic B subunit of coagulation factor XIII (FXIII), which is a tetrameric complex of 2 A and 2 B subunits. When the FXIII-B dimer dissociates in the presence of Ca 2+ , the remaining FXIII-A dimer becomes catalytically active. FXIII is the last zymogen activated in the coagulation cascade, and functions to regulate platelet adhesion and to catalyze fibrin crosslinks—thus stabi-lizing fibrin clots and ECMs. Besides being in close chromosomal proximity, the F13B and CFH/CFHR genes encode structurally similar proteins and are believed to be related evolutionarily. There is also significant crosstalk between the coagulation and complement systems, both of which are proteolytic cascades that are activated in response to injury. The FXIII-A dimer, which is regulated by FXIII-B, is expressed in various leukocytes and downregulated by the classical complement pathway, but is upregulated by the alternative pathway. At least 6 significant AMD-associated F13B SNPs have been identified, one of which (rs6003) has been linked to neovascular AMD risk.
The other major AMD risk locus, 10q26, contains 3 tightly linked genes: PLEKHA1 , ARMS2 , and HTRA1 ( Figure 4 ). With a reported population attributable risk of 57% for a high-risk allele, this locus may be expected to harbor polymorphisms in a large proportion of AMD patients. The functions of genes in this locus are not restricted to immunity, but also may include maintenance of ECM integrity and cellular homeostatis. Their relationship to AMD pathogenesis, although still under investigation, may be pivotal.
PLEKHA1 (10q26.13)
PLEKHA1 encodes the tandem pleckstrin homology domain-containing protein 1 (TAPP1), which regulates B-cell activation and autoantibody production, suggesting that PLEKHA1 may affect AMD through an immunomodulatory function. The role of TAPP1 in AMD has not yet been explored thoroughly. However, TAPP1 knock-in mice display age-related increases in autoantibody production, with a glomerulonephritis phenotype associated with IgG deposition in kidney. This provides yet another possible link between AMD, immune dysfunction, and renal disease.
ARMS2 (10q26.13)
ARMS2 codes for a secreted protein of unclear function. However, the ARMS2 gene product has been shown to bind several ECM proteins, including fibulin 6 (hemicentin-1), fibronectin-1, and elastin microfibril interface-located protein 2, which implies an important role for ARMS2 in ECM maintenance. ARMS2 is expressed in multiple tissues, including the retina, where it has been localized to the choroidal intercapillary pillars in human eyes. The ARMS2 A69S risk allele was recently demonstrated to alter the expression of genes involved in ECM maintenance and cell adhesion in the RPE and choroid of human donor eyes (Whitmore SS, et al. IOVS 2012;53:ARVO E-Abstract 6501). Several groups have demonstrated that the ARMS2 gene product localizes to mitochondria, and there is preliminary evidence that oxidative stress reduces the association of ARMS2 protein with the mitochondrial outer membrane (Boulton ME, et al. IOVS 2012;53:ARVO E-Abstract 5301). Mitochondria are essential for cell survival, but are prone to oxidative damage, and mitochondrial dysfunction is proposed to contribute to the age-related pathologic features of the retina. Thus, variants in ARMS2 may modify AMD risk through mitochondria-related pathways, and although the ARMS2 protein function is still not entirely clear, it may be sensitive to oxidative stress.
HTRA1 (10q26.3)
Although there is a lack of definitive evidence identifying the key contributor within the 10q26 region, HTRA1 stands out as the strongest candidate. A SNP associated with higher HTRA1 expression was identified in a Chinese population and was replicated in a cohort from Utah. In addition, HTRA1 expression is increased in patients with PCV. HTRA1 encodes a member of the high temperature requirement A (HtrA) family of proteases, which are highly conserved across prokaryotic and eukaryotic taxa. In bacteria, HtrA is one of the so-called heat shock proteins that cleave misfolded or denatured proteins, and thus HtrA proteins are critical for cell survival after cellular stresses. In higher organisms, HtrA1 also may influence the integrity of connective tissue through its interaction with ECM components. The HTRA1 protein demonstrates elastase activity, which likely causes the Bruch membrane and choroidal lesions observed in HTRA1 -overexpressing mice. The HtrA serine proteases also may regulate the insulin-like growth factor II axis by their specificity to the insulin-like growth factor-binding proteins. The insulin-like growth factor-binding proteins regulate the distribution and levels of insulin-like growth factor II, which is known to promote angiogenesis. A recent sibling correlation study revealed concordance of advanced AMD subtypes in sibling pairs suggesting that the ARMS2 / HTRA1 locus contributes to both forms of advanced AMD, yet imparts a greater risk of CNV than GA as a final outcome. This further substantiates the role of HTRA1 in progression to neovascular AMD, perhaps through its effects on angiogenesis, neuronal survival, or both. Interestingly, the protease activity of human HTRA1 recently was demonstrated in vitro to degrade tau protein aggregates, which have been implicated in Alzheimer disease and other neurodegenerative diseases. Tau immunoreactivity has been reported in the retina, and although it did not seem to change with age, it did seem to be increased in the RPE of eyes with AMD. Considering the observed roles of the HTRA1 protein in the retina and the evolutionarily conserved functions of HtrA proteases, it seems likely that HTRA1 plays a key role in AMD pathogenesis.
AMD is somewhat unique among disorders exhibiting complex inheritance in that just 2 loci show such a major contribution to the attributable risk. The identification of each of these 2 loci represents a major advance in AMD genetics, and not only confirmed known pathologic pathways, but also sparked additional investigations into other functional areas that are relevant to AMD. Many other loci since have been associated with AMD risk, and they may be important contributors to AMD pathophysiologic processes. By combining clinicopathologic observations with the knowledge gained from recent genetic analyses, we can group AMD risk-associated genes into several functional areas that can be examined in future systems approaches: (1) inflammation and immunity, (2) ECM and cell adhesion, (3) lipid and protein metabolism and transport, (4) angiogenesis, and (5) cellular stress and toxicity. Table 2 provides a compendium of the genes associated with AMD risk, grouped by known functions. Included are genes within the 2 major AMD-associated loci, as well as genes with emerging relevance to AMD risk, which are discussed below.
| Symbol | Name | Locus | Function a | Other Functions a | Association Studies b |
|---|---|---|---|---|---|
| A: INFLAMMATION AND IMMUNITY | |||||
| C2 | Complement component 2 | 6p21.3 | Component of the classical complement pathway | ||
| C3 | Complement component 3 | 19p13.3–p13.2 | Activation of the classical, lectin, and alternative complement pathways | ||
| CFB | Complement factor B | 6p21.3 | Component of the alternative pathway; regulates B lymphocyte proliferation | ||
| CFD | Complement factor D | 19p13.3 | Serine protease; component of the alternative complement pathway | ||
| CFH | Complement factor H | 1q32 | Regulator of the alternative complement pathway | ||
| CFHR1-5 | Complement factor H-related 1-5 | 1q32–1q31.3 | Modulate CFH activity; involved in complement regulation; may be involved in lipid metabolism | C | |
| CFI | Complement factor I | 4q25 | Serine protease; regulates the complement cascade | ||
| CX3CR1 | Chemokine (C-X3-C motif) receptor 1 | 3p21.3 | Receptor for fractalkine, a chemokine involved in leukocyte adhesion and migration | B, E | |
| F13B | Coagulation factor XIII, B polypeptide | 1q31–q32.1 | Evolutionarily related to CFH ; may interact with the complement cascade | B | |
| HLA-C | Major histocompatibility complex, class I, C | 6p21.3 | Regulates natural killer and T-cell activation | ||
| IL8 | Interleukin 8 | 4q13–q21 | Chemoattractant and mediator of inflammation | D | |
| MMP9 | Matrix metallopeptidase 9 | 20q11.2–q13.1 | Interacts with various immunomodulatory molecules; inflammatory biomarker in atherosclerosis | B, D | |
| PLEKHA1 | Pleckstrin homology domain containing, family A (phosphoinositide binding specific) member 1 | 10q26.13 | Regulates B-cell activation and autoantibody production | ||
| RORA | RAR-related orphan receptor α | 15q22.2 | May mediate inflammatory and immune responses | C, D, E | |
| SERPING1 | Serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 | 11q12–q13.1 | Regulates complement activation | ||
| TLR3 | Toll-like receptor 3 | 4q35 | Activation of innate immunity in response to dsRNA | E | |
| TLR4 | Toll-like receptor 4 | 9q33.1 | Activation of innate immunity in response to lipopolysaccharides | E | |
| VLDLR | Very low density lipoprotein receptor | 9p24 | Mediates angiogenesis via inflammation | C, D, E | |
| B: EXTRACELLULAR MATRIX AND CELL ADHESION | |||||
| ACE | Angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 | 17q23.3 | Binds integrins; may modulate cell–ECM interactions | D, E | |
| ARMS2 | Age-related maculopathy susceptibility 2 | 10q26.13 | Component of the ECM; may be involved in matrix function | E | |
| COL8A1 | Collagen, type VIII, α 1 | 3q12.3 | α I chain of type VIII collagen, a major component of specialized ECMs | D | |
| COL10A1 | Collagen, type X, α 1 | 6q21–q22 | α I chain of type X collagen expressed in chondrocytes; proposed to be involved in ECM integrity | D | |
| CST3 | Cystatin C | 20p11.21 | Extracellular inhibitor of cysteine proteases | D, E | |
| CX3CR1 | Chemokine (C-X3-C motif) receptor 1 | 3p21.3 | Receptor for fractalkine (chemokine involved in leukocyte adhesion and migration) | A, E | |
| F13B | Coagulation factor XIII, B polypeptide | 1q31–q32.1 | Catalyzes fibrin crosslinks and stabilizes fibrin clots in the ECM | A | |
| FBLN5 | Fibulin 5 | 14q32.1 | Secreted ECM protein | D | |
| HMCN1 | Hemicentin (fibulin 6) | 1q25.3–q31.1 | Elastin-associated component of the ECM | ||
| HTRA1 | HtrA serine peptidase 1 | 10q26.3 | Serine protease; cleaves fibulin 5 | D, E | |
| MMP9 | Matrix metallopeptidase 9 | 20q11.2–q13.1 | Degrades type IV and V collagens | A, D | |
| ROBO1 | Roundabout, axon guidance receptor, homolog 1 | 3p12 | Regulates axon guidance and neuronal migration | ||
| TIMP3 | Tissue inhibitor of metalloproteinase 3 | 22q12.3 | Matrix metalloproteinase inhibitor | D | |
| C: LIPID/PROTEIN METABOLISM AND TRANSPORT | |||||
| ABCA1 | ATP-binding cassette, subfamily A (ABC1), member 1 | 9q31.1 | Cholesterol efflux pump | ||
| ABCA4 | ATP-binding cassette subfamily A member 4 | 1p22 | Retina-specific lipid transporter; functions in clearing all- trans -retinal | E | |
| APOE | Apolipoprotein E | 19q13.2 | Essential for the catabolism of triglyceride-rich lipoproteins by the liver; maintains normal lipid homeostasis | E | |
| CETP | Cholesteryl ester transfer protein, plasma | 16q21 | Transfers cholesteryl esters between lipoproteins | ||
| CFHR1-5 | Complement factor H-related 1–5 | 1q32–1q31.3 | Secreted members of the CFH family; associate with lipoproteins and may be involved in lipid metabolism | A | |
| CYP24A1 | Cytochrome P450, family 24, subfamily A peptide 1 | 20q13 | Involved in cholecalciferol (vitamin D3) degradation | E | |
| ELOVL4 | ELOVL fatty acid elongase 4 | 6q14 | Involved in fatty acid biosynthesis. | ||
| FADS1-3 | Fatty acid desaturases 1–3 | 11q12.2–q13.1 | Fatty acid synthesis | ||
| LIPC | Hepatic lipase | 15q21–q23 | Receptor-mediated lipoprotein uptake | ||
| LPL | Lipoprotein lipase | 8p22 | Receptor-mediated lipoprotein uptake | ||
| LRP6 | Low density lipoprotein receptor-related protein 6 | 12p13.2 | Receptor-mediated endocytosis of lipoprotein and protein ligands | D | |
| RORA | RAR-related orphan receptor α | 15q22.2 | Nuclear hormone receptor; implicated in cholesterol/lipid metabolism | A, D, E | |
| VLDLR | Very low-density lipoprotein receptor | 9p24 | VLDLR-triglyceride metabolism | A, D, E | |
| D: ANGIOGENESIS | |||||
| ACE | Angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 | 17q23.3 | Catalyzes the conversion of angiotensin 1 into angiotensin II, which is involved in angiogenesis regulation | B, E | |
| COL10A1 | Collagen, type X, α 1 | 6q21–q22 | α I chain of type X collagen, which may be pro-angiogenic | B | |
| COL8A1 | Collagen, type VIII, α 1 | 3q12.3 | α I chain of type VIII collagen, which is expressed by vascular smooth muscle cells and may be proangiogenic | B | 177 |
| CST3 | Cystatin C | 20p11.21 | Extracellular inhibitor of cathepsin, which regulates angiogenesis | B, E | |
| FBLN5 | Fibulin 5 | 14q32.1 | Secreted ECM protein; promotes adhesion of endothelial cells; involved in vascular development and remodeling | B | |
| GDF6 | Growth differentiation factor 6 | 8q22.1 | Secreted member of the TGF-β and bone morphogenic protein families; mediates angiogenic regulation by HTRA1 | ||
| HTRA1 | HtrA serine peptidase 1; high temperature requirement factor A1 | 10q26.3 | Regulates angiogenesis via GDF6 and IGFB5 | B, E | |
| IL8 | Interleukin 8 | 4q13–q21 | Chemoattractant and potent angiogenic factor | A | |
| LRP6 | Low density lipoprotein receptor-related protein 6 | 12p13.2 | Coreceptor for Wnt/β-catenin signaling pathway, which is required for vascular development; antagonized by PEDF | C | |
| MMP9 | Matrix metallopeptidase 9 | 20q11.2–q13.1 | Mediates angiogenesis | A, B | |
| RORA | RAR-related orphan receptor α | 15q22.2 | Nuclear hormone receptor; implicated in angiogenesis | A, C, E | |
| SERPINF1 | Serpin peptidase inhibitor, clade F | 17p13.3 | Encodes PEDF, a potent angiogenesis inhibitor | ||
| TIMP3 | Tissue inhibitor of metalloproteinase 3 | 22q12.3 | Matrix metalloproteinase inhibitor; angiogenesis inhibitor; involved in vascular remodeling | B | |
| VEGFA | Vascular endothelial growth factor A | 6p12 | Vascular permeability factor, endothelial cell mitogen, and angiogenic factor | ||
| VLDLR | Very low-density lipoprotein receptor | 9p24 | Mediates angiogenesis via inflammation | A, C, E | |
| E: CELLULAR STRESS AND TOXICITY | |||||
| ABCA4 | ATP-binding cassette sub-family A member 4 | 1p22 | Retina-specific ABC transporter ABCR; prevents toxic accumulation of A2E | C | |
| ACE | Angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 | 17q23.3 | Catalyzes the conversion of angiotensin 1 into angiotensin II; angiotensin signaling mediates cellular stress | B, D | |
| APOE | Apolipoprotein E | 19q13.2 | Involved in the cellular stress response; proteolytic fragments are neurotoxic | C | |
| ARMS2 | Age-related maculopathy susceptibility 2 | 10q26.13 | Unclear function; mitochondrial expression is modulated by oxidative stress | B | |
| CST3 | Cystatin C | 20p11.21 | Involved in the cellular stress response; inhibits amyloid-beta deposition and toxicity; implicated in ECM remodeling in retinal degeneration | B, D | |
| CX3CR1 | Chemokine (C-X3-C motif) receptor 1 | 3p21.3 | Involved in the modulation of apoptosis in response to cellular stress | A, B | |
| CYP24A1 | Cytochrome P450, family 24, subfamily A peptide 1 | 20q13 | Involved in cholecalciferol (vitamin D3) degradation | C | |
| GSTM1 | Glutathione S-transferase mu 1 | 1p13.3 | Xenobiotic metabolism and detoxification | ||
| GSTP1 | Glutathione S-transferase pi 1 | 11q13 | Xenobiotic metabolism and detoxification | ||
| GSTT1 | Glutathione S-transferase τ 1 | 22q11.23 | Xenobiotic metabolism and detoxification | ||
| HTRA1 | HtrA serine peptidase 1; high temperature requirement factor A1 | 10q26.3 | Involved in the cellular stress response | B, D | |
| RORA | RAR-related orphan receptor α | 15q22.2 | Involved in the cellular stress response | A, C, D | |
| SOD2 | Superoxide dismutase 2, mitochondrial | 6q25.3 | Xenobiotic metabolism and detoxification | ||
| TF | Transferrin | 3q22.1 | Maintains iron homeostasis | ||
| TLR3 | Toll-like receptor 3 | 4q35 | Involved in the cellular stress response to dsRNA | A | |
| TLR4 | Toll-like receptor 4 | 9q33.1 | Involved in the cellular response to lipopolysaccharides and environmental stress | A | |
| VLDLR | Very low-density lipoprotein receptor | 9p24 | May protect against oxidative stress-induced photoreceptor death | A, C, D | |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


