FIGURE 9.1. Schematic diagram showing how diabetes in conjunction with hypertension and hyperlipidemia causes alteration of the BRB, leading to DME. Both increased levels of growth factors and proteinases, as well as leukostasis/inflammation result in endothelial cell junction breakdown. Several steps of this cascade can be used as therapeutic targets to inhibit DME.
The key pathological event that occurs in macular edema is increased vascular permeability. Vascular permeability has shown to be initiated by various vaso-permeable agents such as VEGF, TNF, Interleukin, and Ang2 that are upregulated in DME.31,32 The other important and nascent area of research in terms of DME is inflammation. TNF, interleukins, and adhesion molecules such as Inter cellular adhesion molecule (ICAM) and Vascular cell adhesion molecule (VCAM) can attract inflammatory cells to the retina. This phenomenon is called retinal leukostasis. These activated inflammatory cells can release various soluble chemokines that can induce vasopermeability. Alternatively, these inflammatory cells can breach the endothelial barrier through the process called diapedesis leading to the pathogenesis of DME.33–36 The detailed information of the importance of BRB, inflammation, growth factors, and the potential therapeutic targets of DME is discussed further below.
Composition of the Blood-Retinal Barrier
The blood-retinal barrier is one of most important ocular architectures, which is involved in the tight regulation and maintenance of the fluid electrolyte balance in the retina. Basically, the BRB partition the neural retina from the circulation by acting as a barrier for the molecules that can enter. Breakdown of this barrier contributes to the fluid accumulation in the retinal disorders including DME. The inner BRB is made of the endothelial cells in the inner retina, and the outer BRB by retinal pigment epithelial (RPE) cells on the Bruch membrane between the fenestrated choroidal vessels and the outer retina.28,29
The endothelial cells form a very tight monolayer, which is helped by the adherens and the tight junction proteins. Adherens junctions are formed through the interactions between cadherins, a family of functionally related transmembrane glycoproteins, which mediate calcium-dependent, cell-cell adhesion.37 The important adherent junctional protein is the VE-cadherin (cadherin-5). VE-cadherin proteins are expressed specifically by the endothelial cells, which are implicated in a number of endothelial cell functions. The most important function of VE-cadherin is the maintenance of the endothelial cell barrier function.38 Endothelial monolayer permeability is regulated by VE-cadherin through the interaction of its cytoplasmic tail with cytoplasmic proteins called catenins. β-Catenin, plakoglobin (γ-catenin), and α-catenin bring out the attachment of the cadherin complex with the actin cytoskeleton. The attachment of cadherin to the actin cytoskeleton is required for the formation of a restrictive endothelial monolayer.39,40 A fourth catenin, p120, associates with the cadherin cytoplasmic domain just proximal to the membrane, a region termed the juxtamembrane domain (JMD).41,42 The phosphorylation state of p120 plays an important role in regulating cadherin function. Studies have indicated that molecules such as thrombin, histamine, and importantly VEGF decrease endothelial barrier function mediated through the phosphorylation mechanism of catenins. Also, the retinal endothelial cells and the pigmented epithelial cells are tightly connected through a network of intercellular tight junctional proteins that regulate fluid flux. It has been estimated that around 40 tight junctional proteins in the peripheral cytoplasm and apical plasma membrane provide a fine control of influx of water and solutes.43 The principal proteins found in endothelial tight junctions are occludin, claudin-5, and JAMs, which create a tight seal so that water-soluble molecules cannot easily penetrate between the cells. These transmembrane proteins have linkages to the actin cytoskeletal proteins such as zona occludin-1 (ZO-1) and zona occludin-2 (ZO-2). 1 details the function of some important junctional proteins involved in the maintenance of BRB.
Also the BRB integrity is maintained tightly by the presence of pericytes in the endothelium. Pericytes cover the apical side of the lumen and are separated by the matrix from the endothelial cells. Brain microvascular pericytes have been shown to send out processes through the lamina, which help to communicate with endothelial cells. It is estimated that the endothelial: pericyte ratio is 1:1 in the retina, and any decrease in the pericyte cell number leads to increased vascular permeability. In diabetes, there is an increased pericyte loss, which leads to an increased leakage of the retinal capillaries.44 Also, the ECM acts as a foundation for the endothelial cells to form a monolayer. The basement membrane is mainly composed of collagen IV, fibronectin, and laminin that interact with endothelial cells and aid in adhesiveness to the matrix.45 Degradation of the matrix results in endothelial cells migration and also in the loss of barrier function.
Alteration of Blood-Retinal Barrier in Diabetes
The disruption of BRB in diabetes involves numerous factors and events within the vascular lumen and in the retinal parenchyma. Prominent among them is the VEGF, which was initially called as vascular permeability factor. The inflammatory factors such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) have also shown to alter the vascular permeability through various molecular mechanisms. Other growth factors and cytokines including hepatocyte growth factor (HGF), FGF, insulin-like growth factor-1 (IGF-1), and histamine have shown to alter the retinal vascular permeability.46–48
It has been proposed that the factors that contribute to DME alter the starling equilibrium in the retina (role of hydrostatic and oncotic forces (the so-called Starling forces) in the movement of fluid across capillary membranes), leading to diminished fluid reabsorption capacity resulting in fluid retention within the microvessel lumens. One important example is that the diabetic patients associated with macroalbuminuria will have decreased oncotic pressure due to hypoalbuminemia, which will diminish the fluid retention capacity of the vessel lumens.49 Those diabetic patients with persistent macroalbuminuria due to poorly controlled diabetes will have severe macular edema. Also, diabetes drugs such as glitazones have been implicated in the aggravation of macular edema due to its fluid retention property.50
The important mechanism that leads to vision loss in DME due to altered BRB involves (a) light scattering, (b) disruption of retinal ionic balances, and (c) impaired cell-cell communication that involves not only the retinal microvascular cells but also the retinal neuronal cells.46 The details on the physical aspects of the visual-optometric functional impairment and its molecular details are still not known but the association of vascular leakage with the vision impairment has been well established.51 The importance of the fluid accumulation or the edema involves multiple and/or prolonged insult to the macula. Importantly, the RPE cells act as the major fluid-absorbing sponge in the retina, but in prolonged insults, they lose their ability, thus leading to the progression of macular edema and subsequent vision loss.52 These final events include the dysfunction of retinal glial and neuronal cells, and therefore the functional restoration of those vascular and neuronal cells may provide a better therapy for maintaining vision.
PROTEINASES IN DME
The vitreous level of MMP9 was higher in diabetic subjects with DR than with the diabetic subjects without DR. This study shows that MMPs must have a potential role in the pathogenesis of DR.53 Furthermore, in animal model of diabetes, both MMP2 and MMP9 are elevated, which are involved in the alteration of blood-retinal barrier.54 Another form of MMP called MT1-MMP, which is a membrane-bound MMP, is also increased along with MMP2 in the diabetic animals, and concomitant to this, there is an increased apoptosis of pericytes in the diabetic retina when compared to the normal retina. This may further accelerate BRB alteration in the diabetic state. We have shown that retinal vascular permeability was significantly increased in rats following 2 weeks of diabetes, coincident with a decrease of VE-cadherin expression. This increased vascular permeability could be inhibited with an MMP inhibitor (Figs. 9.2–9.6). AGE increases a loss of VE-Cadherin, which is prevented by MMP inhibitor, suggesting that MMP has a direct role in the alteration of endothelial monolayer.55 These observations suggest a possible mechanism by which diabetes contributes to BRB breakdown through proteolytic degradation of VE-cadherin. This may indicate a role for extracellular proteinases in alteration of the BRB seen in diabetic retinopathy.55 High glucose can activate many soluble mediators such as AGE, ROS, inflammatory cytokines, which can increase MMP expression and activity in the diabetic state. In a recent work, the oxidative stress has been implicated to have an influence in the MMP2 and MT1-MMP expressions and activities.56 The role of MMP-9 is implicated in the alteration of barrier function, which is shown to be mediated by TGF-β.57 Studies have hinted that diabetes causes retinal inflammation, which unleashes a sequelae of events resulting in the vascular leakage. Retinal inflammation attracts increased leukocytes to the retina, which then bind to the vascular endothelium. The binding of leukocyte to the endothelial cells activates cellular proteases such as elastase, which may clip off VE-cadherin and its associated protein from the cell surface, resulting in endothelial monolayer alteration.58
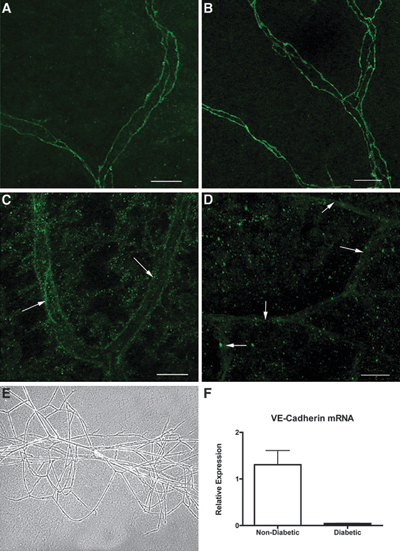
FIGURE 9.2. VE-cadherin protein and mRNA are decreased in the retinal vessels of diabetic rats. Representative retinal whole mounts from control and diabetic rats stained with anti–VE-cadherin antibody and examined by confocal microscopy. VE-cadherin expression was evident at the interfaces between adjacent endothelial cells in control nondiabetic animals (A,B), while it was mostly obliterated from the microvessels in the diabetic retina at 2 weeks (C) and 8 weeks (D) of diabetes. Images shown are reconstructions of a series of Z-stacks of retinal blood vessel segments. Vascular networks were isolated from whole retinas of diabetic and nondiabetic animals, as described in research design and methods. A phase-contrast image of such a network is shown in (E). Total RNA was isolated, and semiquantitative real-time RT-PCR was performed. Vessels from diabetic animals demonstrated less VE-cadherin mRNA than vessels from non–diabetic controls (F). *Significantly less than nondiabetic controls (p = 0.0002). n = 3 animals for staining and PCR analysis. Bar = 25 μm. (Reproduced from Navaratna, et al. Diabetes. 2007;56:2380–2387, with permission.)
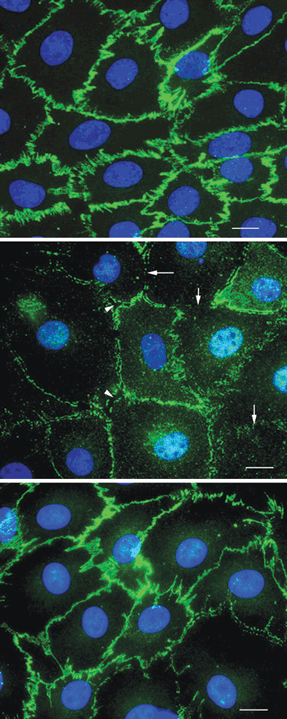
Figure 9.3. AGE stimulation alters endothelial cell morphology and cell surface expression of VE-cadherin. Representative images of bovine retinal endothelial cells cultured in serum-free media for 48 hours under various treatment conditions, fixed, and stained for VE-cadherin. The treatment conditions were serum-free media alone (A), AGE-BSA (50 μg/mL) (B), and AGE-BSA (50 μg/mL) with BB-94 (2 μmol/L) (C). Disruption of the continuous pattern of VE-cadherin staining (arrows) and the presence of open gaps between adjacent cells (arrowheads) are seen in cultures treated with AGE-BSA. n = 3 cultures from two separate experiments. Bar = 10 μm. (Reproduced from Navaratna, et al. Diabetes. 2007;56:2380–2387, with permission.)
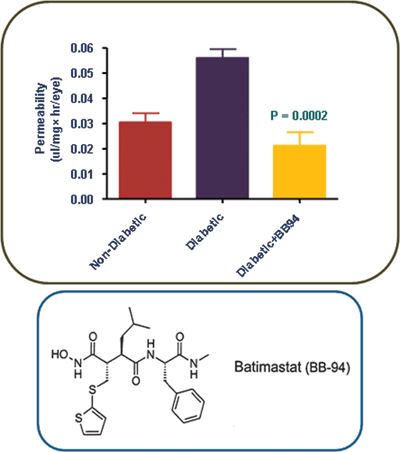
Figure 9.4. MMP inhibitor blocks retinal vascular permeability in the diabetic rat. Diabetic rats received an intraperitoneal injection of the MMP inhibitor BB-94 (1 mg/kg) daily for 2 weeks. Retinal vascular permeability was quantitated using the Evans blue assay. The permeability of the retinal vasculature was nearly twofold lower in BB-94–injected diabetic rats compared with vehicle-injected diabetic rats, Diabetic versus nondiabetic, p < 0.01; diabetic + MMP inhibitor versus diabetic, p < 0.01; nondiabetic versus diabetic + MMP inhibitor, p > 0.05. (Reproduced by permission from Navaratna, et al. Diabetes. 2007;56:2380–2387.).
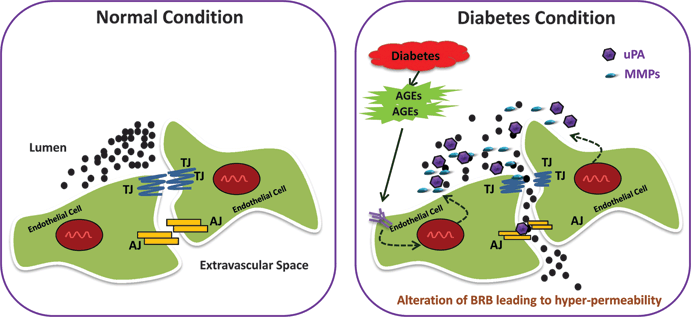
Figure 9.5. Alteration of the BRB in diabetes. Under normal conditions, the inner BRB is maintained by the cell junctional proteins (tight and adherens junctions) between the endothelial cells. In diabetes, hyperglycemia increases expression of proteinases (matrix metalloproteinases and urokinase) that proteolytically degrade cell junctional proteins, thus allowing molecules to come out of the lumen to the extracellular space.
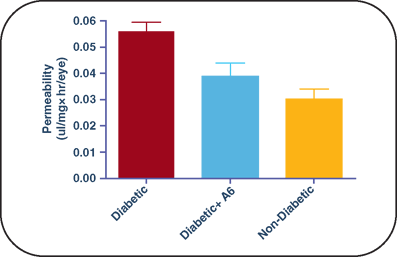
FIGURE 9.6. Å6 treatment prevents microvascular permeability and loss of VE-cadherin in the diabetic retina. Retinal vascular permeability was assessed using the Evans blue dye technique. The permeability of the retinal vasculature in Å6-injected diabetic rats is comparable to that of noninjected nondiabetic controls. Diabetic versus diabetic + Å6 or nondiabetic, p < 0.001; diabetic + Å6 versus nondiabetic, p > 0.05. (Reproduced by permission from FASEB J. 2008;22:3310–3317).
ROLE OF INFLAMMATION IN DME
Recent studies reveal that inflammatory cytokines play a major role in the pathogenesis of DME. Many cytokines such as VEGF, ICAM-1, IL-6, and MCP-1 are increased in the vitreous of DME patients when compared with diabetic patients without any form of diabetic retinopathy or nondiabetic subjects. Among those inflammatory mediators, VEGF and ICAM-1 had a stronger influence on the severity of DME.32 IL-1β is a potential inflammatory cytokine, which causes severe alteration to the retinal permeability, the effects of which are inhibited by TGF-β.59 TNF-α is an important cytokine that is increased in diabetes. IL-β and TNF-α have shown to alter the retinal barrier after exposure. TNF-α and IL-1β induced changes in the tight junctional protein adhesion property of endothelial cells, leading to extensive cellular infiltration leading to inflammation and hemorrhage.60–62 The adhesion molecules such as ICAM-1 can also attract leukocytes to the diabetic retina, and these leukocytes penetrate the retinal barrier through a process called diapedesis and thus cause a BRB breakdown. In the hyperglycemic state, the increased formation of AGE mediates increased retinal leukostasis and upregulation of integrins, which are involved in the leukocyte adhesion.63
In an animal model of experimental diabetes, retinal leukostasis was increased within a few weeks of diabetes and the retina displayed classical signs of diabetic retinopathy including increased capillary permeability and loss of endothelial cells and pericytes, resulting in acellular capillaries. Diabetic mice lacking CD18 and ICAM-1 had very few adherent leukocytes in the retina and did not display the sign and symptoms of DR, indicating that the inflammatory markers ICAM-1 and CD18 are crucial for retinal leukostasis.64 TNF-α is one of the crucial mediators of retinal leukostasis, and studies have shown that mice lacking TNF-α had 80% reduction in retinal leukostasis even in the presence of VEGF (Box 9.1)65.
BOX 9.1 Infl ammation and DME
Increased retinal leukostasis has shown to alter the retinal endothelial function, resulting in increased vascular permeability in diabetes. This process might lead to the development of capillary nonperfusion, endothelial cell damage, and vascular leakage leading to DME. Leukostasis has also been indicated in the increased secretion of Tumor necrosis factor-α (TNF-α) and Interleukin-1 β (IL-1β), which are proinflammatory cytokines. Intravitreous injection of TNF-α and IL-1β causes BRB breakdown and inflammation. It has been proposed that high glucose levels induce leukocyte-endothelial cell (EC) adhesion by upregulating cell surface adhesion molecules. Inflammatory mediators such as PKC, TNF-α, and nuclear factor-κB (NF-κB) have been linked to glucose-mediated leukocyte-EC adhesion. Leukocytes in the diabetic environment release a higher amount of reactive oxygen species as compared with cells in a physiologic environment. This results in generation of toxic superoxide radicals and proteolytic enzymes. The release of these reactive metabolites and the trapped leukocytes in the endothelium will damage the vessel wall and cause leakage of intravascular contents into the surrounding tissue, leading to the development of DME. Currently various steroids and nonsteroidal anti-inflammatory drugs are being tested in clinical trials for the treatment of DME.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


