of Ocular Angiogenesis
Shozo Sonoda • Parameswaran G. Sreekumar • Ram Kannan • David R. Hinton
Introduction
Physiologic angiogenesis is carefully regulated by a critical balance between endogenous stimulators and inhibitors. However, this balance can be tipped toward a more angiogenic phenotype under pathological conditions such as inflammation, ischemia, oxidant injury, and tumor growth. While most attention in studies of ocular angiogenesis has been focused on angiogenic stimulators, the focus of this chapter is on endogenous inhibitors of angiogenesis and how they may be regulated or utilized to prevent or treat ocular angiogenic disorders. The emphasis of this chapter is on those endogenous inhibitors of angiogenesis that have been shown to modulate angiogenesis in preclinical ocular models of disease and those that have the greatest potential for clinical application.
The number and variety of endogenous inhibitors of angiogenesis, which have been identified over the past 2 decades, are surprisingly large. An unusual feature of many of these endogenous inhibitors of angiogenesis is that they are cleavage products of larger proteins without any activity related to angiogenesis or are antiangiogenic splice variants of proteins involved in proangiogenic pathways. Endogenous inhibitors of angiogenesis can be classified into several defined categories based on their derivation and activity. These include extracellular matrix (ECM)–derived angiogenesis inhibitors (Table 8.1); growth factors, soluble growth factor receptors, and cytokines (Table 8.2); fragments of blood coagulation factors (Table 8.3); and a fourth category of other molecules having potent antiangiogenic activity (Table 8.4). The matrix-derived factors include those derived from larger ECM proteins such as collagens (arresten, canstatin, endostatin, tumstatin), fibronectin (anastellin), and perlecan (endorepellin) or are found as intact ECM molecules such as Fibulin 3 and 5, and thrombospondin (TSP). The prominent members of the second category encompassing growth factors, soluble receptors, and cytokines are pigment epithelium–derived factor (PEDF), a splice variant of vascular endothelial growth factor (VEGF165b), transforming growth factor beta (TGF-β), and soluble extracellular domains of the receptors for VEGF (sFlt1) and the angiopoietins (sTie2). The third category includes fragments of blood coagulation, namely angiostatin, antithrombin III, and prothrombin kringle-2 and platelet factor-4. The fourth category includes a diverse group of angiogenesis inhibitors including apoptosis signal-regulating kinase-1 interacting protein-1 (AIP1), cleavage products of prolactin (PRL) (vasoinhibins), a fragment of matrix metalloproteinase-2 (PEX), troponin I, vasostatin, 2-methoxyestradiol, chondromodulin-1, tissue inhibitors of metalloproteinases, and tryptophan tRNA synthetase.
Table 8.1.
Matrix-derived endogenous angiogenesis inhibitors
| Endogenous Inhibitor | Size in kDa | Production Site | Mechanism of Action |
|---|---|---|---|
| Arresten | 26 | Derived from the COOH-terminal of NC1 domain of the α1 chain of type IV collagen.1 | Inhibition of endothelial cell proliferation, migration, tube formation, blood vessel formation.1 Inhibition of FAK/c-Raf/MEK/ERK1/2/p38 MAPK activation in ECs. Inhibit HIF1α expression.2,3 |
| Canstatin | 24 | Derived from the COOH-terminal of NC1 domain of the α2 chain of type IV collagen.4 | Inhibition of endothelial cell migration, tube formation, proliferation, and induction of apoptosis.4 Canstatin inhibits the phosphorylation of multiple signaling molecules in endothelial cells.3 It binds to integrins αvβ3 and αvβ5, which initiates apoptotic pathways.4,5,7 |
| Endorepellin | 85 | A basement membrane heparin sulfate proteoglycan C-terminal domain V of human perlecan8 | Inhibition of endothelial cell migration, collagen-induced endothelial tube morphogenesis, and vessel growth in the chorioallantoic membrane, and Matrigel plug assays. Endorepellin binds endostatin and counteracts its antiangiogenic effects.8 Angiostatic effects in vivo are mediated by a specific interaction of endorepellin with the α2β1 integrin receptor.9 Endorepellin triggers a signaling cascade, leading to disruption of the actin cytoskeleton.10 |
| Endostatin | 20 | Conditioned media of murine hemangioendothelioma cells.11 NC1 domains of Basement membrane collagens.12,13 | Inhibition of migration of endothelial cells by blocking downstream signaling of α5β1 integrin (Src kinase, RhoA GTPase, Ras, Raf).2 Endostatin also binds to HSPGs that are involved in growth factor signaling.14 These events promote disassembly of the actin cytoskeleton, disorders in cell-matrix interactions, and decrease in endothelial mobility.15 Endostatin inhibits proliferation of endothelial cells, inducing their apoptosis through inhibition of cyclin D1. Endostatin induces autophagy by upregulating and complexing Beclin 1, Bcl-2, and Bcl-x (L).16 |
| Anastellin | 29 | Derived from the first type III repeat of fibronectin | Anastellin blocks serum-dependent proliferation of endothelial cells by suppressing Ras activation and preventing ERK activation and subsequent cell proliferation.17 Inhibits endothelial cell migration.18 |
| Fibulin 3 and 5 | 50–60 | Vascular smooth muscle, endothelial cells | Fibulin inhibits endothelial cell tubulogenesis, proliferation, and migration.19 It reduces angiogenic sprouting stimulated by bFGF, inhibits matrix metalloproteinase expression and activity, and stimulates tissue inhibitor of metalloproteinase expression. It prevents angiogenesis and vessel infiltration into bFGF-supplemented Matrigel plugs implanted in mice.20 |
| TSP | 145 | ECM glycoprotein synthesized by endothelial cells, epithelial cells, smooth muscle cells, and macrophages. | TSP-1 inhibits endothelial cell migration, adhesion, and proliferation and induces apoptosis. TSP-1 activates TGF-β. TSP-1 antagonizes VEGF-mediated survival signaling, inhibits VEGF mobilization by MMP9, and directly binds VEGF. TSP-1 antagonizes VEGF-driven Akt survival, signaling in part through the recruitment of Fyn to membrane domains containing CD36.21 |
| Tumstatin | 28 | Purified from MMP-degraded basement membrane preparations from kidney, placenta, and testis.22 | Inhibits formation of new blood vessels in Matrigel plug assay by inducing apoptosis.22 It inhibits endothelial proliferation. Through the interaction with αvβ3integrin, tumstatin inhibits activation of FAK, P13K, protein kinase B/Akt, and prevents dissociation of eukaryotic initiation factor 4E protein from 4E-BP1, leading to inhibition of Cap-dependent protein synthesis.23 Binding of tumstatin to integrin αvβ3 is dependent on the PTEN/Akt pathway.24 |
Table 8.2.
Growth factors, soluble receptors, and cytokines as endogenous angiogenesis inhibitors
| Endogenous Inhibitor | Size in kDa | Origin | Mechanism of Action |
|---|---|---|---|
| PEDF | 50 | Ocular sites of synthesis include RPE, endothelial cells, corneal epithelial cells, lens epithelial cells, ciliary epithelial cells, and retinal ganglion cells. | Inhibits angiogenesis by induction of FasL and apoptosis of endothelial cells.25 Inhibits vascular permeability and vascular leakage by preventing blood retinal breakdown through tight junction restoration. Inhibits inflammatory molecules. MAPK, JNK, and p38 influence endothelial cell apoptosis by modulating c-FLIP or caspase activity in the presence of PEDF.26–28 |
| VEGF165b | 38 (nonglycosylated) | Distal splice variant of VEGF165. In primary RPE cultures, VEGF165b is major form of VEGF. | Inhibits VEGF- and hypoxia-induced angiogenesis, and VEGF-induced cell migration and proliferation in vitro. Inhibits oxygen-induced ROP. IGF1 and TNFα treatment favors proximal splice site selection (increasing VEGF165), whereas TGF-β1 favored distal splice site selection (DSS), increasing VEGFb levels. TGF-β1-induced DSS selection was prevented by inhibition of p38 MAPK and the CLK/sty (CDC-like kinase, CLK1) splicing factor kinase family, but not ERK1/2.29 |
| TGF-β | 25 | Widely expressed in epithelial, endothelial, hematopoietic, neural, and certain mesenchymal cells.30 | Contact with pericytes and endothelial cells activate TGF-β to inhibit endothelial cell migration, proliferation and differentiation of pericytes. TGF-β treatment switches VEGF splicing toward antiangiogenic isoforms.29 |
| Soluble Flt-1 | 180 | Extracellular domain of Flt1 receptor. Expressed in endothelial cells, monocytes, and corneal epithelial cells.31 | It includes the extracellular ligand-binding domain but not the transmembrane and intracellular domains; it is secreted (hence “soluble”) and antagonizes VEGF and PlGF by binding and preventing their interaction with endothelial VEGF receptors.31 sFlt-1 blocks VEGF-induced cell proliferation and migration. |
| Soluble Tie2 | 160 | Extracellular domain of Tie2. Expressed by vascular endothelial cells | Angiopoietins, the natural ligands of Tie2, modulate Tie2-dependent signaling, which in turn regulates the survival and apoptosis of endothelial cells, controls vascular permeability, and regulates the capillary sprouting that occurs during normal angiogenesis.34 sTie2 binds angiopoietins to prevent binding to receptor. |
Table 8.3.
Fragments of blood coagulation as endogenous angiogenesis inhibitors
| Endogenous Inhibitor | Size in kDa | Origin | Mechanism of Action |
|---|---|---|---|
| Angiostatin | 38 kDa | Internal fragment of plasminogen | Inhibits endothelial cell proliferation.35,36 Inhibits the F(1)-F(0) ATP synthase expressed on endothelial cell surface, allowing the intracellular pH to drop, triggering apoptotic events in the endothelial cells.37,38 Angiostatin induces apoptosis by downregulating mitochondrial Bcl-2.39 |
| Antithrombin III | 53–55 | Cleaved form of antithrombin III | Inhibits endothelial cell proliferation.40 |
| Prothrombin kringle-2 | 22 | ||
| Platelet factor-4 | 70 aminoacid | Released from alpha-granules of activated platelets.41 | Inhibits blood vessel proliferation in chicken chorioallantoic membrane. Inhibits angiogenesis by associating directly with FGF-2, inhibiting its dimerization and blocking FGF-2 binding to endothelial cells.42 |
Table 8.4.
Other endogenous angiogenesis inhibitors
| Endogenous Inhibitor | Size in kDa | Origin | Mechanism of Action |
|---|---|---|---|
| AIP1 | 70–75 | vascular endothelial cells, epithelial cells | AIP1 through its Ras GTPase-activating protein activity inhibits Ras-mediated cell survival signaling, causing cell growth inhibition.43 AIP1 regulates apoptosis by mediating activation of ASK1.44 AIP1 binds to VEGFR2-P13K complex and inhibits the VEGFR2-dependent angiogenesis.45 Inhibits VEGF-induced migration and tube formation. |
| Vasoinhibins (PRL) | 16 | Generated by proteolytic cleavage of PRL, growth hormone, and placental lactogen in pituitary gland. Vasoinhibins occur naturally in the eye.46 | Vasoinhibins interfere with ocular angiogenesis by blocking several endothelial functions and their actions mediated by the inactivation of endothelial nitric oxide synthase. They inhibit endothelial cell attachment to laminin and reduce endothelial growth induced by FGF-2.47 |
| PEX | 210 amino acid fragment | Noncatalytic COOH-terminal hemopexin-like domain of MMP2. | PEX prevents binding of MMP-2 to integrin αvβ3, inhibiting proteolytic activity on the cell surface and disrupting angiogenesis. Inhibits cell invasion, formation of capillary-like structures.48 |
| Troponin I | 21 | Cartilage-derived | Inhibits endothelial cell proliferation and angiogenesis.49 |
| Vasostatin | aa 1–180 | NH2-domain of calreticulin inclusive of aa 1–180 | Inhibits angiogenesis by inhibiting endothelial cells’ proliferation and inducing apoptosis.50,51 |
| 2-Methoxyestradiol | 0.28 | Endogenous 2ME2 is synthesized by the hydroxylation at the 2-position of estradiol and subsequent O-methylation by catechol-O-methyltransferase | Destabilization of microtubules with a block in nuclear accumulation and activity of HIF-1α, leading to a significant reduction in VEGF. It has ability to bind to the colchicine-binding site of tubulin and the inhibition of superoxide dismutase enzymatic activity.52 2ME2 inhibits growth and sprouting of endothelial tubular formations. Inhibits angiogenesis induced by bFGF.53 |
| Chondromodulin-1 | 25 | Cartilage-specific NC1 matrix protein | Inhibits tube morphogenesis of retinal endothelial cells and DNA synthesis.54,55 |
| Tissue inhibitors of metalloproteinases | 21–28 | Astrocytes, endothelial cells, epithelial cells, fibroblasts | Suppress MMP activity and ECM turnover. TIMP-2 inhibits cell proliferation and angiogenesis.56,57 |
| Tryptophan tRNA synthetase (T2-TrpRS) | 43–48 | Proteolysis or alternative splicing of TrpRS.58 | T1-TrpRS and T2-TrpRS block VEGF-induced migration of human umbilical vein endothelial cells. TrpRS blocks VEGF-stimulated angiogenesis in in vivo murine matrigel assay.59 A recombinant form of a COOH-terminal fragment of TrpRS is a potent antagonist of VEGF-induced retinal angiogenesis.58 T2-TrpRS inhibits pathologic neovascularization and facilitates physiological revascularization of ischemic tissue.60 |
Endogenous Inhibitors of Angiogenesis as Mediators of Avascularity in The Eye
A unique feature of the eye is that there are naturally avascular structures, which require a lack of vascularity in order to attain normal function with optical clarity; these include the cornea and the fovea. Interestingly, it is the high expression of endogenous antiangiogenic inhibitors that is thought to generate and maintain this avascular environment. For the cornea, high expression of sFlt-1 is thought to maintain avascularity,33 while for the fovea, high expression of PEDF in the ganglion cell layer is thought to generate the avascular area.91
Inhibition of Angiogenesis in Different Ocular Models
Pathological ocular angiogenesis is a key component of many blinding ocular diseases such as age-related macular degeneration (AMD), diabetic retinopathy, retinopathy of prematurity (ROP), and corneal angiogenesis. Animal models of ocular angiogenesis are among the most frequently utilized in the evaluation of antiangiogenic agents because of their ease of analysis and reproducibility. In fact, many of the endogenous inhibitors of angiogenesis were first studied or were evaluated in most detail in ocular models of angiogenesis. Our laboratory has shown that endogenous angiogenesis inhibitors such as PEDF and TSP-1 are differentially regulated in the murine ROP model in a strain-dependent manner,92 suggesting that strain must be carefully considered when evaluating angiogenesis inhibition in animal models. A detailed table listing the preclinical assessment of endogenous angiogenic inhibitors in various models of ocular angiogenesis is provided (Table 8.5). In most preclinical animal studies, the endogenous angiogenic inhibitors are delivered as recombinant proteins/peptides by various routes (intraperitoneal, intravenous, subconjunctival, intravitreous, subretinal), by overexpression using viral vectors (adenovirus, adenoassociated virus, lentivirus) delivered systemically, intravitreally, or subretinally, or by use of nanoparticles (Fig. 8.1).
Table 8.5.
Inhibition of angiogenesis in ocular models of neovascularization
| Species | Pharmacologic Approach | Route of Administration | Ocular Condition/Disease | Outcome |
|---|---|---|---|---|
| Endostatin | ||||
| Mouse | rh endostatin | Intra peritoneal | Laser-induced CNV | CNV lesions were almost undetectable in endostatin-injected animals. Endostatin at physiological levels inhibited induced angiogenesis in vivo and reduced vascular leakage.61 |
| Mouse | AAV-endostatin | Subconjunctival | Corneal neovascularization | AAV-endostatin significantly inhibited corneal neovascularization induced by silver nitrate.62 |
| Mouse | AAV-endostatin | Subretinal | Retinal neovascularization | AAV-endostatin reduced retinal neovascularization, vascular permeability, and retinal detachment in VEGF transgenic mouse.63 |
| Mouse | AAV-endostatin | Subretinal | ROP | Significantly inhibited ischemia-induced neovascularization.64 |
| Mouse | AAV-endostatin | Intravenous | Laser-induced CNV | ~10-fold higher endostatin serum levels and nearly complete prevention of CNV. A strong inverse correlation between endostatin serum level and area of CNV was observed.65 |
| VEGF 165b | ||||
| Mouse | rhVEGF165b peptide | Intravitreous | ROP | Inhibited the percentage area of retinal neovascularization from 23% to 12% and increased normal vascular areas from 62% to 74%.66 |
| PEDF | ||||
| Mouse | Ad5-PEDF/Ad35-PEDF | Intravitreous | Laser-induced CNV | Ad35 better than Ad5 vector with respect to durability and level of transgene expression. PEDF expression from an Ad35.PEDF vector was able to inhibit CNV lesion growth by greater than 80% at Day 42 as compared to the no-injection control.67 |
| RCS rat | SIV-hPEDF/SIV-hFGF2 | Subretinal | Retinitis pigmentosa | SIV-hPEDF or SIV-hFGF-2 significantly delayed the histological degeneration. Delays were synergistically and significantly pronounced by combined injection of SIV-hPEDF and SIV-hFGF-2.68 |
| Mouse | Recombinant PEDF/PEDF peptide/PEDF-nanospheres | Intravitreous | Retinal ischemia-reperfusion | PEDF prevented approx. 44% of the cell death in the RGC layer. PEDF82–121 peptide was as effective as full-length PEDF when injected as either a free peptide or delivered in PLGA nanospheres.69 |
| Mouse | Recombinant PEDF | Intravitreous | AGE-induced diabetes | 50% decrease in blood retinal breakdown and vascular permeability in PEDF-treated diabetic groups.70 |
| Rat | Recombinant PEDF | Intravitreous | OIR | Most potent effect of PEDF was observed at 48 h after injection, with the retinal vascular permeability in PEDF-treated eyes decreasing to 56% of the PBS control (downregulation of VEGF, VEGF receptor-2, MCP-1, TNF-α, and ICAM-1).28 |
| Rat | rPEDF | Intravitreous | STZ-diabetes | PEDF prevented VEGF-induced endothelial permeability and blood-retina-barrier breakdown, at least partially by blocking the disorganization of tight junction proteins.28 |
| Mouse | PEDF peptide | Intravitreous | Nonproliferative diabetic retinopathy | Reduced VEGF-induced vascular permeability by 84%.71 |
| Mouse | Recombinant PEDF | Intravitreous | Nonproliferative diabetic retinopathy | Inhibited VEGF-induced vascular permeability by 96%.71 |
| Mouse | AAV-PEDF | Intravitreous | Retinal ischemia –reperfusion injury | Significantly increased cell survival after ischemia-reperfusion injury of the retina.72 |
| RCS rat/rds mice | Simian lentiviral vector-PEDF | Subretinal | Retinitis pigmentosa | Significant protection of loss of photoreceptor cells in regions of gene transfer with fewer TUNEL-positive cells. PEDF-treated eyes retained sensitivity to light flash.73 |
| Mouse | rAAV2-PEDF | Intravitreous | ROP | Decrease in number of neovascular tufts; overall vasculature pattern appeared similar to that in the normal animal. Endothelial cell counts in PEDF-treated eyes were reduced by 74% compared with controls.74 |
| Mouse | AdrPEDF | Intravitreous | CNV | Prominent immunoreactivity for PEDF in RPE with strong inhibition of CNV.75 |
| Mouse | Recombinant PEDF | Intravenous | ROP | Vascular tufts were reduced or not observed. At highest dose (22.4 μg/d) of PEDF, there was 67% inhibition of neovascularization.76 |
| Rat | Recombinant PEDF/peptide | Hydron pellets implanted into the avascular rat cornea | Corneal neovascularization | Significantly inhibited corneal neovascularization.77 |
| Mouse | AdPEDF.11 | Periocular | Laser-induced CNV | Periocular injection of AdPEDF.11 strongly suppressed CNV.78 |
| Mouse | AdPEDF.11 | Periocular | ROP | Periocular injection of AdPEDF.11 did not inhibit retinal neovascularization because retinal PEDF levels were 10-fold lower than levels of PEDF in the choroid.78 |
| Mouse | AAV-PEDF | Intravitreal PEDF transfection | Inherited glaucoma | Potent reduction of retinal ganglion cell loss and vision decline with decrease in TNF, IL-18, and gliosis.79 |
| RCS Rat | SIV-hPEDF | subretinal | Retinitis pigmentosa | Protected photoreceptors from cell death by inhibiting the nuclear translocation of apoptosis-inducing factor with upregulation of Bcl-2.80 |
| Rat | AdPEDF.11 | Intravitreous | Light-induced photoreceptor cell death | Increased photoreceptor cell survival by inhibition of light-induced apoptotic processes.81 |
| TSP-1 | ||||
| Mouse | Wispostatin-1 (TSP-1 peptide repeat) | Intravitreous | Corneal micropocket (bFGF) and laser induced CNV | Abolished bFGF-induced neovascularization in corneal micropocket assay. Significant inhibition of CNV was also found with peptide treatment.82 |
| Rat | rhTSP-1 and TSP-1 peptide | Intravitreous | ROP | Decrease in retinal neovascularization with platelet-derived TSP-1 or rh-TSP-1.83 |
| Soluble Flt-1 | ||||
| Rat | Ad-sFlt-1 | Intramuscular | Laser-induced CNV | Increased serum levels of sFlt-1. Less prominent vessel formation with decreased fibroblast proliferation and decreased inflammatory cell infiltration.84 |
| Rat | Ad-HD-sFlt-1 | Intravitreous | OIR | 60% inhibition in retinal neovascularization. Average endothelial cell count was reduced by 80%.85 |
| Rat (Torii SDT) | Ad-sFlt-1 | Subretinal | Spontaneous Diabetes | Decrease in avascular area, hyperfluorescence, and arterial narrowing 30 weeks after vector administration. No adverse effects were observed.86 |
| Rat | Ad-sFlt-1 | Intravitreous | ROP | 98% decrease in peak retinal neovascularization. No significant difference in retinal vessel number detected in oxygen-injured and normoxic groups at p28.87 |
| Mouse | sFlt-1-Fc (recombinant fusion protein) | Intravitreous | ROP | 37% inhibition of retinal neovascularization. Retinal vasculature appeared relatively intact.88 |
| Mouse | AAV-sFlt-1/AdsFlt-1 | Intravitreous | ROP | Reduction in the number of neovascular endothelial cells by 56% and 52% for adenovirus and AAV vectors, respectively.89 |
| Tryptophanyl-tRNA synthetases | ||||
| Mouse | rh mini TrpRS/T2-TrpRS | Intravitreous | Physiological angiogenesis in the neonatal retina | Complete inhibition of the outer network was observed in 28% of mini TrpRS (5 pmol)-treated eyes. The smaller T2-TrpRS variant was far more potent, with dose-dependent activity; 14% were completely inhibited after treatment with 1 pmol of T2-TrpRS, 40% after treatment with 2.5 pmol and 69% after 5 pmol.58 |
| Mouse | T2-TrpRS | Intravitreous | OIR | A strong, dose-dependent angiostatic effect of T2-TrpRS on pathologic neovascularization in the OIR model was observed. Injection of 1.25 μg/eye T2-TrpRS resulted in nearly complete inhibition of neovascular tuft formation. T2-TrpRS also inhibited pathologic neovascularization and reduced tuft formation by more than 75%.60 |
| Soluble Tie2 | ||||
| Mouse | AAV-Tie2 receptor | Intramuscular | ROP | Treatment inhibited retinal neovascularization by 47%.90 |
| Mouse | AAV-Tie2 receptor | Intramuscular | Laser-induced CNV | Treatment significantly reduced the incidence and extent of fluorescein leakage from CNV lesions by 52% and 36%, with 45% reduction in CNV area.90 |
| Mouse | sTie2-Fc (recombinant fusion proteins) | Intravitreous | ROP | Retinal neovascularization at P17 reduced in 92% of animals. The average magnitude of inhibition compared with IgG control was 23%.88 |
Supported by: The Arnold and Mabel Beckman Foundation, National Institutes for Health Grants (EY01545, EY03040), and a grant to the Department of Ophthalmology by Research to Prevent Blindness.
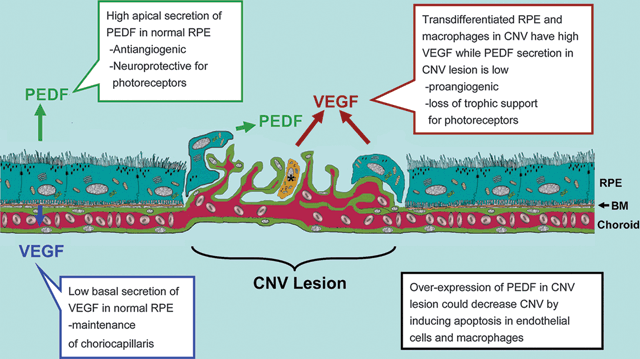
FIGURE 8.1. Diagram illustrating the balance between PEDF and VEGF in the normal RPE monolayer and in CNV. The left side of the diagram shows that RPE cells normally secrete high levels of PEDF from their apical surface, which may provide photoreceptor neuroprotection. Normal RPE cells also secrete low levels of VEGF from their basal surface, which may allow for maintenance of the choriocapillaris. Under conditions of neovascularization (CNV), choroidal endothelial cells break through Bruch membrane and form subretinal neovascular channels. Transdifferentiated RPE and macrophages (indicated with an asterisk) within the lesion secrete significantly higher amounts of VEGF and much less PEDF, thereby altering the VEGF/PEDF ratio and creating a proangiogenic microenvironment.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


