6 Pediatric Otolaryngology Section Editor Contributors • The airway is relatively narrower and more tenuous in children. • The potential for airway emergency is high. • Many conditions causing respiratory distress in infants resolve spontaneously with growth. The pediatric airway is proportionally smaller than the adult: the tongue is relatively larger and more anterior, the soft palate descends lower, the adenoid is larger, the epiglottis is omega shaped, larger, and more acutely angled toward the glottis; the cricoid ring is narrower, the trachea is shorter and narrower, the surrounding soft tissue is looser, and cartilaginous structures are less rigid. Thus the pediatric airway is more prone to compromise by infection, inflammation, neoplasia, and normal breathing. Foreign body aspiration may be a life-threatening emergency. An aspirated solid or semisolid object may lodge in the airway. If the object is large enough to cause nearly complete obstruction of the airway, asphyxia may rapidly cause death. Infants are at risk for foreign body aspiration because of their tendency to put everything in their mouths and because of immature chewing. Children may be asymptomatic. If present, physical findings may include stridor, fixed wheeze, or diminished breath sounds. If obstruction is severe, cyanosis may occur. • Stridor: Harsh high-pitched sound of turbulent airflow past partial obstruction in upper airway • Stertor: Low-pitched, snorting sound resulting from partial nasal/nasopharyngeal/hypopharyngeal obstruction • Wheezing: A continuous whistling or musical sound on expiration from a small bronchiole constriction For subjective assessment of respiratory distress, see Table 6.1. Indications for intubation for airway compromise include PaO2 <60 mm Hg with FiO2 >0.6 (without cyanotic heart disease), PaCO2 >50% (acute, unresponsive to other intervention), actual or impending obstruction, neuromuscular weakness (maximum negative inspiratory pressure over −20 cm H2O, vital capacity <12–15 mL/kg), and an absent cough/gag reflex. • Supralaryngeal • Laryngeal • Tracheobronchial A thorough history of respiratory distress (cyanosis, apnea, dyspnea, retractions, grunting) including age of onset, frequency, degree, and rate of progression, ameliorating and exacerbating factors (including positional), as well as feeding difficulties and fevers should be elicited. Difficult delivery, history of prematurity, and postpartum complications (asphyxia, duration of intubation) should be noted. On the physical exam, note respiratory rate, nasal flaring, intercostal and supraclavicular retraction, gasping, or respiratory fatigue. Examine the mouth using a tongue blade, unless epiglottitis is suspected. Auscultate the chest and neck. Note change in breathing when positioning child upright, supine, prone, and on each side. Visualize airway with flexible laryngoscope, unless epiglottitis is suspected. More severe cases may require bronchoscopy and possibly esophagoscopy. Bronchoscopy (rigid or flexible) may be both diagnostic and therapeutic (see A1 in Appendix A). Upright anteroposterior (AP) and lateral soft tissue x-rays of the airway (i deally on full inspiration with head in extension) best evaluate the upper airway. Additionally, AP and lateral chest x-rays can detect extrinsic compression or evaluate for evidence of airway foreign body. If child has feeding difficulties, obtain a barium esophagram. Magnetic resonance imaging (MRI) with MR angiography (MRA) is a good alternative to angiography for vascular malformations. Obtain gastric emptying scans and esophageal pH monitoring if reflux is suspected. For foreign body aspiration, employ posteroanterior inspiratory and expiratory chest radiography to look for hyperinflation, computed tomography (CT), or fluoroscopy. Studies may be false-negative; if index of suspicion remains high, bronchoscopy in the OR is indicated. Although pulmonary function testing with a flow-volume loop can help distinguish inspiratory versus expiratory as well as intrathoracic versus extrathoracic obstruction, the test requires a cooperative subject and is not often feasible in children <6 years. Polysomnography is very helpful in the evaluation of possible pediatric sleep-related respiratory disorders; a differentiation between obstructive versus central apnea can be obtained. This is highly recommended in children with a history of neurologic disorder. Acute choking, with respiratory failure associated with airway foreign body obstruction, may be successfully treated at the scene using standard first aid techniques such as the Heimlich maneuver, back blows, and abdominal thrusts. Even in less urgent settings, expeditious removal of airway foreign bodies is recommended and a workup may be performed. Definitive management will, of course, depend upon the specific diagnosis. But in general terms, for the child with airway compromise continuous monitoring with pulse oximetry is necessary. Supplemental humidified oxygen, racemic epinephrine, or heliox may be implemented. Systemic steroids are often employed. Reflux medications such as lansoprazole (Prevacid, Takeda Pharmaceutical Company, Deerfield, IL), to decrease gastric acid production, as well as metoclopramide (Reglan, Schwarz Pharma, Inc., Milwaukee, WI), to promote gastric emptying, can help with reflux irritation of the airway. Upper respiratory infections should be empirically treated as an inpatient with ceftriaxone 75 mg/kg/day intravenously (IV), or comparable antibiotic covering Streptococcus pneumoniae, Streptococcus pyogenes, and Haemophilus influenzae. Again, definitive management will, of course, depend upon the specific diagnosis. In cases of severe compromise, pediatric tracheotomy may be necessary. In laryngomalacia, redundant laryngeal tissue can be excised, and aryepiglottic folds can be divided using endoscopic scissors or CO2 laser. However, many cases of laryngomalacia may be managed with observation (see Chapter 6.2). Subglottic stenosis can be dilated, or a cricoid split or cartilage graft reconstruction performed (see Chapter 6.7). Obstructing masses should be removed, if possible. Subglottic hemangioma management is discussed in Chapter 6.13. The child should be monitored closely overnight in case of bleeding or edema compromising airway. Oxygen saturation should be monitored. Specific conditions are discussed in detail in the following chapters. Outcome depends on the diagnosis and management. Child can be followed with the usual well-child checks, and immunizations should be kept up to date.
6.1 Pediatric Airway Evaluation and Management
Key Features
Clinical
Signs and Symptoms
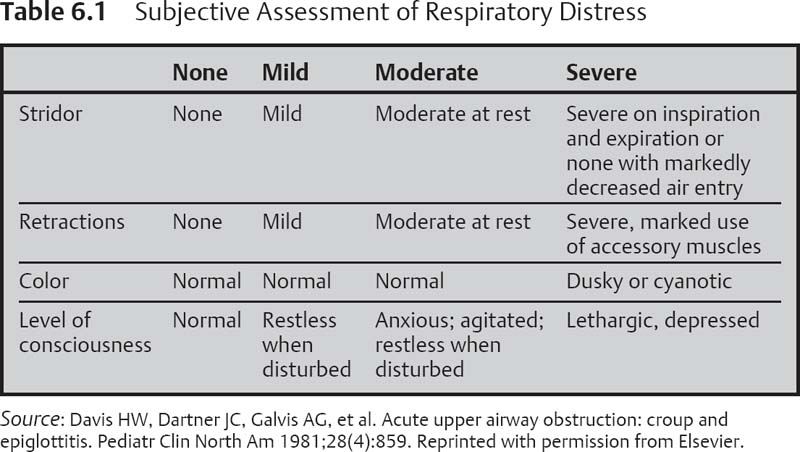
 Inspiratory stridor signifies supraglottic obstruction.
Inspiratory stridor signifies supraglottic obstruction.
 Biphasic stridor signifies glottic or subglottic obstruction.
Biphasic stridor signifies glottic or subglottic obstruction.
 Expiratory stridor signifies tracheal or large bronchial compression.
Expiratory stridor signifies tracheal or large bronchial compression.
Differential Diagnosis
 Adenoid hypertrophy
Adenoid hypertrophy
 Macroglossia
Macroglossia
 Mass (nasopharyngeal, base of tongue)
Mass (nasopharyngeal, base of tongue)
 Choanal atresia
Choanal atresia
 Foreign body
Foreign body
 Cellulitis
Cellulitis
 Neck/pharyngeal abscess
Neck/pharyngeal abscess
 Laryngomalacia (most common overall cause of pediatric stridor)
Laryngomalacia (most common overall cause of pediatric stridor)
 Vocal fold paralysis (unilateral often is iatrogenic or trauma related, bilateral is often due to central nervous system [CNS] lesion or dysfunction)
Vocal fold paralysis (unilateral often is iatrogenic or trauma related, bilateral is often due to central nervous system [CNS] lesion or dysfunction)
 Laryngeal web
Laryngeal web
 Subglottic stenosis or hemangioma
Subglottic stenosis or hemangioma
 Papillomata
Papillomata
 Laryngeal cleft
Laryngeal cleft
 Viral croup
Viral croup
 Epiglottitis
Epiglottitis
 Gastric reflux
Gastric reflux
 Tracheomalacia
Tracheomalacia
 Bronchomalacia
Bronchomalacia
 Vascular ring
Vascular ring
 Airway foreign body
Airway foreign body
 Bronchitis
Bronchitis
 Bronchiolitis
Bronchiolitis
 Tracheoesophageal fistula (TEF)
Tracheoesophageal fistula (TEF)
Evaluation
History
Physical Exam
Imaging
Other Tests
Treatment Options
Medical
Surgical
Outcome and Follow-Up
ICD-9 Codes
 Stenosis
Stenosis786.1 | Stridor |
748.3 | Other anomalies of larynx, trachea, and bronchus |
Further Reading
Myer CM, Cotton RT, Shott SR. Pediatric Airway: An Interdisciplinary Approach. Philadelphia, PA: JB Lippincott; 1995
Wetmore RF, Muntz HR, McGill TJ, et al. Pediatric Otolaryngology: Principles and Practice Pathways. Stuttgart/New York: Thieme; 2000
6.2 Laryngomalacia
Key Features
• Most common cause of stridor in infants (accounts for ~75% of infantile stridor).
• Often self-limited; most patients are symptom-free by 12 to 24 months of age.
• Treatment for 90% of cases is expectant observation.
• Its etiology is unknown.
Laryngomalacia is a temporary physiologic dysfunction due to abnormal flaccidity of laryngeal tissues or incoordination of supralaryngeal structures.
Epidemiology
Laryngomalacia is the most common congenital airway abnormality. Overall, laryngomalacia accounts for 60% of cases of chronic laryngeal stridor. It is more common among males than females (2:1). Concomitant airway abnormalities are found in 12 to 37% of patients. Comorbidities, including prematurity, cardiovascular malformation, and neurologic and congenital or chromosomal abnormalities, are present in ~41% of patients.
Clinical
Signs and Symptoms
Most commonly, patients present with intermittent inspiratory stridor that is relieved by neck extension and a prone position. Stridor is exacerbated by agitation. In extreme cases, patients become cyanotic, have a poor oral intake, have chest retractions, and develop pectus excavatum.
Differential Diagnosis
Other causes of stridor in the early pediatric age group are
• Unilateral or bilateral vocal fold paralysis
• Laryngeal cleft
• Choanal atresia
• Airway hemangioma
• Laryngeal web
• Airway foreign body
• Acquired or congenital subglottic stenosis
• Craniofacial anomalies
• Glottic cysts
• Laryngeal reflux
• Saccular cyst
• Tracheomalacia
• Papillomatosis
Acid-reflux disorders (see Chapter 4.5) have been documented in up to 80% of patients with laryngomalacia.
Evaluation
Physical Exam
An examination of any child with a possible breathing problem should discern if there is an oxygenation problem, and if so, an oxygen requirement. In the physical examination, one should assess for the location of a possible obstruction and include auscultation, inspection for chest retraction, assessment for cyanosis and other anomalies such as micrognathia.
To diagnose laryngomalacia and assess for other upper airway abnormalities, direct flexible endoscopic examination during respiration must be performed. Inward collapse of supraglottic structures are visualized on inspiration. The vocal folds are normal in appearance and motility.
Direct laryngoscopy and bronchoscopy in the operating room is the definitive evaluation.
Imaging
Radiologic examination is not generally necessary. However, if there is concern about dysphagia, barium swallow is helpful to assess esophageal function and to assess for evidence of vascular rings or other obstructive lesions or evidence of TEF.
Pathology
Histologically, submucosal edema and lymphatic dilatation are seen. Mechanisms of obstruction have been described by numerous authors. Chen and Hollinger considered the occurrence of two or more synchronously as causal in airway obstruction. These factors include an inward collapse of aryepiglottic folds, an elongated epiglottis (flaccid) curled on itself, anterior and medial collapsing movements of the arytenoid cartilages, posterior and inferior displacement of the epiglottis, short aryepiglottic folds, and an overly acute angle of epiglottis.
Treatment Options
Medical
If an infant has good progress, which is indicated by adequate weight gain and normal development, then surgical therapy is not necessary. Instead, supportive care can be the mainstay of treatment.
Surgical
In one series of 985 patients with laryngomalacia, 12% required surgical intervention. Patients who should be considered for surgical management are those with severe stridor and failure to thrive, obstructive apnea, weight loss, severe chest deformity, cyanotic attacks, pulmonary hypertension, or cor pulmonale. Supraglottoplasty is performed using carbon dioxide laser or laryngeal microscissors, or other cold instruments such as pediatric ethmoid thru-cutting forceps. Most commonly, surgery involves removal of the prolapsing aryepiglottic fold with cuneiform cartilage or division of tight, short aryepiglottic fold. Unilateral supraglottoplasty has been advocated by some to reduce the risk of supraglottic stenosis. If epiglottis is displaced posteriorly, an epiglottopexy can be performed.
Potential complications include continued airway obstruction and posterior stenosis. In the event of continued airway obstruction, a tracheotomy may be necessary until the child “outgrows” laryngomalacia. The use of postoperative antibiotics has not been well evaluated in the literature and is controversial. Antireflux medications are used routinely.
Outcome and Follow-Up
Supraglottoplasty relieves symptoms of airway obstruction in 90% of patients. Most infants require only a short hospital stay (1–3 days).
ICD-9 Code
748.3 | Laryngomalacia |
Further Reading
Belmont JR, Grundfast K. Congenital laryngeal stridor (laryngomalacia): etiologic factors and associated disorders. Ann Otol Rhinol Laryngol 1984;93(5 Pt 1):430–437
Cotton RT, Reilly JS. Congenital malformations of the larynx. In: Bluestone CD, Stool SE, eds. Pediatric Otolaryngology. 2nd ed. Philadelphia, PA: WB Saunders; 1990:1121–1128
Tunkel DE, Zalzal GH. Stridor in infants and children: ambulatory evaluation and operative diagnosis. Clin Pediatr (Phila) 1992;31(1):48–55
Zalzal GH. Stridor and airway compromise. Pediatr Clin North Am 1989;36(6):1389–1402
6.3 Bilateral Vocal Fold Paralysis
Key Features
• Bilateral vocal fold paralysis is the second most common cause of infantile stridor.
• It typically requires a tracheotomy for maintenance of airway until vocal fold mobility returns, or definitive airway surgery is performed.
• Acquired causes are most common, even in infants.
Bilateral vocal fold paralysis (BVFP) is a potentially lethal problem requiring aggressive management, typically tracheotomy, at least in the short term. Depending on the cause, spontaneous recovery can occur. Recovery, however, is generally a slow process, taking up to a year. A variety of surgical approaches to widen the airway have been described. Usually, however, the voice quality is degraded when there is an intervention to enlarge/improve the laryngeal airway.
Epidemiology
Twenty-five percent of BVFP is congenital. Most of the remaining cases are late presentations of central lesions. Acquired cases are most likely to occur as a result of surgery in the chest or from forceps delivery or infections.
Clinical
Signs
Vocal fold immobility can be seen using flexible laryngoscopy in the clinic. Stridor is a sign of BVFP.
Symptoms
• Stridor
• Typically a normal voice, episodic respiratory distress (e.g., with upper respiratory tract infections)
• Weak cough; aspiration if underlying cause is a neural lesion above the nodose ganglion
Differential Diagnosis
• Vocal fold fixation
• Laryngeal mass
• Laryngeal web
Evaluation
Physical Exam
Assess for stridor, respiratory effort and rate, color, and weight (plot on a growth chart). Flexible laryngoscopy is mandatory (it may show twitching of cords with respiration; it typically shows paramedian vocal folds).
Imaging
An MRI should scan from skull base to mediastinum (to image the complete course of laryngeal nerves).
Other Tests
Tests depend on the clinical scenario. For example, consider audiometry if a CNS disorder is present; consider a biopsy of nonvascular laryngeal mass lesion, if present.
Pathology
Congenital Causes
• Laryngeal anomalies, malformations
• CNS anomalies: Arnold-Chiari malformation, hydrocephalus, encephalocele, leukodystrophy, cerebral dysgenesis
• Peripheral neuropathy: neonatal myasthenia gravis, benign congenital hypotonia, Werdnig-Hoffmann disease, Charcot-Marie-Tooth disease, arthrogryposis, viral neuropathy
• Birth trauma, including perinatal hypoxia
Acquired Causes
• Sequelae of cardiac or esophageal surgery
• Neoplasia
• Infections
• Trauma
Treatment Options
Medical
No medical treatment options are available other than supportive care initially to maintain ventilation and oxygenation during definitive treatment planning.
Surgical
• Tracheotomy and monitoring for spontaneous recovery (minimum of 1 year)
• Arytenoidopexy, endoscopic vocal fold lateralization procedures
 Lateralization sutures passed through the vocal process and thyroid ala lateralize the vocal fold without mucosal destruction
Lateralization sutures passed through the vocal process and thyroid ala lateralize the vocal fold without mucosal destruction
• Arytenoidectomy
 External approach (lateral cervical or via laryngofissure)
External approach (lateral cervical or via laryngofissure)
 CO2 laser via endoscopic approach
CO2 laser via endoscopic approach
• Posterior cartilage graft
• Laryngeal reinnervation – not widely used in children
Complications
Accidental tracheotomy displacement can be fatal in the early recovery period. Other complications related to laryngeal airway surgeries include dysphonia, aspiration in 4 to 6%, and dyspnea in 3 to 8% of patients undergoing arytenoid procedures. Other procedures have not been studied enough to ascertain their complication rates. Assessment for aspiration should be done preoperatively, as posterior glottic expansion surgery can increase risk of aspiration.
Outcome and Follow-Up
Initially, the patient is monitored in an intensive care unit (ICU) setting with continuous pulse oximetry. Placement of “stay sutures” for any pediatric tracheotomy is mandatory. These two Prolene sutures, placed through cartilage adjacent to the tracheal opening at the time of surgery and secured to the neck skin with Steri-Strips, greatly facilitate the replacement of a tracheotomy tube into the airway if there is accidental displacement. To help prevent displacement, the tracheotomy appliance should be secured to the skin with four sutures as well as the umbilical neck tie.
Overall, arytenoidopexy and arytenoidectomy yield high rates of successful decannulation. These patients require long-term follow-up.
ICD-9 Codes
478.34 | Bilateral vocal fold paralysis |
786.1 | Stridor |
Further Reading
Brigger MT, Hartnick CJ. Surgery for pediatric vocal fold paralysis: a meta-analysis. Otolaryngol Head Neck Surg 2002;126(4):349–355
De Jong AL, Friedman EM. Vocal fold paralysis. In: Wetmore RF, Muntz HR, McGill TJ, et al, eds. Pediatric Otolaryngology: Principles and Practice Pathways. Stuttgart/New York: Thieme; 2000:787–800
Myer CM, Cotton RT, Shott SR. The Pediatric Airway: An Interdisciplinary Approach. Philadelphia, PA: JB Lippincott;1995
6.4 Laryngeal Clefts
Key Features
• Laryngeal clefts are a rare congenital anomaly.
• They are frequently associated with other anomalies.
• Clinical presentation varies with the extent of the cleft. The patient may have problems with the airway, feeding, and voice.
• Most clefts are short, but complete laryngotracheoesophageal clefts have a mortality rate greater than 90%.
Laryngeal clefts represent a rare cause of stridor. Failed fusion of the posterior cricoid lamina and incomplete development of the tracheoesophageal septum results in a laryngeal cleft, an abnormal communication of the larynx and esophagus. Laryngeal clefts are usually sporadic nonsyndromic congenital abnormalities. Most are associated with other nonsyndromic congenital abnormalities including TEF, esophageal atresia, congenital heart disease, cleft lip and palate, micrognathia, glossoptosis, laryngomalacia, gastrointestinal and genitourinary anomalies. Rarely, they are attributable to a specific syndrome [Opitz-Frias and Pallister-Hall syndrome, VATER (v ertebrae, a nus, t rachea, e sophagus, and r enal) Association].
Epidemiology
Laryngeal clefts are rare. Congenital anomalies of the larynx are found in only 0.5% of the population, and clinically symptomatic laryngeal clefts constitute only 0.3 to 0.5% of all congenital laryngeal anomalies. Males are more likely to be affected than females (3:1). Thirty percent of laryngeal clefts are associated with maternal polyhydramnios. TEF is present in ~25% of patients with a laryngeal cleft and is associated with a higher failure rate of surgical repair.
Clinical
Signs and Symptoms
There are no pathognomonic findings. Clinical symptoms depend on the extent of the cleft. Small clefts, whose anatomic involvement is limited to the interarytenoid musculature, present with stridor and feeding problems. There may be coughing, choking, stridor, aspiration pneumonias, or cyanotic episodes. Occasionally, patients with small laryngeal clefts may be asymptomatic. However, the most severe clefts are accompanied by aphonia, severe upper airway obstruction, and respiratory distress. Stridor occurs due to anterior collapse of posterior supraglottic structures. Cyanosis and stridor are exacerbated with feeding. In utero, polyhydramnios due to impaired swallowing of amniotic fluid by the fetus is associated with laryngeal clefts.
Differential Diagnosis
• Subglottic stenosis
• Laryngomalacia
• Unilateral or bilateral vocal fold paralysis
• Subglottic hemangioma
• TEF
Evaluation
Physical Exam
A full head and neck exam should be performed, including a fiberoptic airway exam. Attention should be directed at possible sources of airway symptoms, such as choanal atresia, craniofacial anomalies, laryngomalacia, and vocal fold motion impairment.
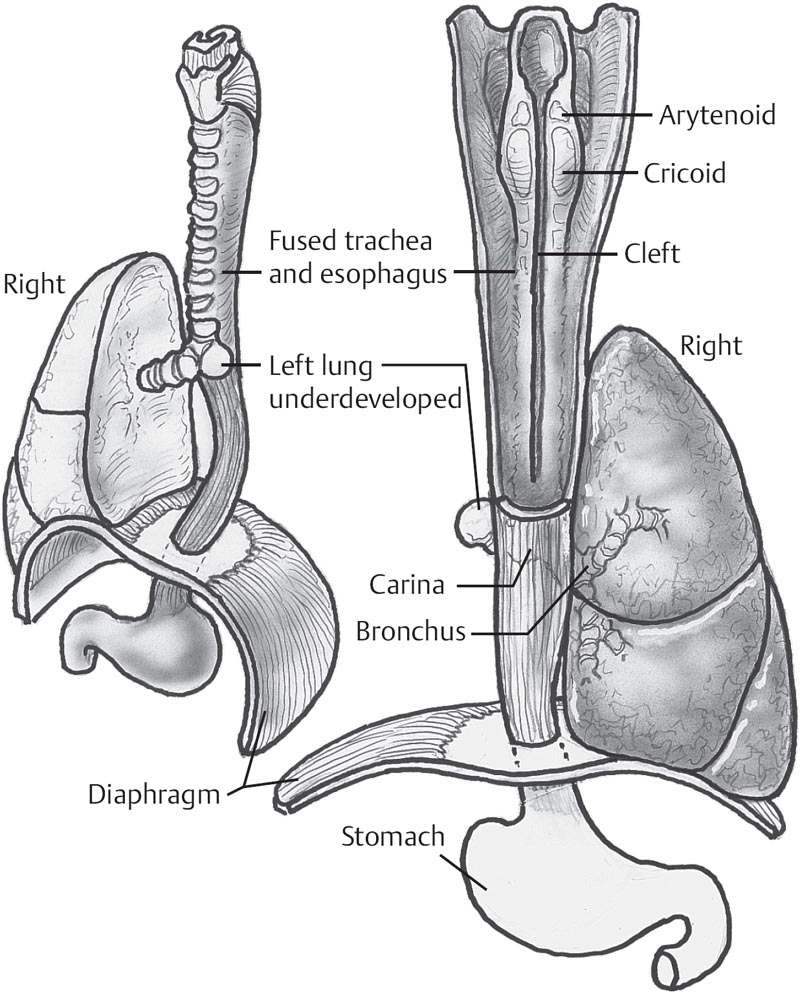
Fig. 6.1 Left anterolateral view and posterior view of type IV laryngotracheoesophageal cleft with left-side pulmonary agenesis and microgastria.
Laryngeal clefts must be visualized. Suspension microlaryngoscopy is used to visualize and palpate a laryngeal cleft. Palpation is performed on the lateral spread of the interarytenoid mucosa. The normal interarytenoid height from the vocal fold is 3 mm, and is severely reduced in laryngeal clefts. Bronchoscopy and esophagoscopy are necessary to adequately assess the airway and to investigate concomitant anomalies (Fig. 6.1).
Imaging
Modified barium swallow and a chest radiograph should be performed. Aspiration with thin liquids is the most common finding. A fiberoptic endoscopic evaluation of swallowing (FEES) is helpful preoperatively.
Pathology
There are several classification schemes in the literature. Because of the rare nature of this disorder, there is no consensus, although the Benjamin/Inglis Classification is commonly used. The classification is based on the inferior extent of cleft:
• Type 1: Interarytenoid soft tissue cleft without extension into cricoid cartilage.
• Type 2: The cleft extends into cricoid cartilage.
• Type 3: The cleft extends through the entire posterior cricoid cartilage.
• Type 4: The cleft extends into the thoracic trachea; it may extend to the carina.
Treatment Options
Medical
Speech and feeding therapy aimed toward decreasing aspiration may be used in conjunction with medical therapy if the cleft is short. For clefts that extend only through interarytenoid musculature but not into cricoid, antireflux therapy in conjunction with thickened diet may be sufficient to control symptoms. However, most clefts require repair. In general, any cleft associated with significant aspiration is ruptured.
Surgical
The surgical approach should be individualized based on the symptoms, other associated findings on airway endoscopy, and type of cleft. The decision for surgical repair initially hinges on the extent of the cleft with respect to cricoid and its relationship with local anatomy. Endoscopic techniques may be feasible for type 1 and same type 2 clefts. However, if the cleft extends into inferior anatomic structures, open surgery is generally necessary. Type 3 and 4 clefts require open repair. This is also an option for patients who fail endoscopic management. Early repair is important to minimize irreversible pulmonary damage due to persistent aspiration.
Laryngeal clefts that extend into cricoid or trachea, without carinal involvement, are accessed via anterior laryngofissure to avoid neurovascular damage. Clefts that extend into the cricoid and involve carina require either a lateral pharyngotomy and lateral thoracotomy or an anterior laryngofissure and median sternotomy. Sternotomy requires cardiopulmonary bypass during the intrathoracic portion and perioperative tracheotomy. Clefts repaired via open approach in young children most often require tracheotomy. This likely is kept in place for an extended period as these patients tend to have substantial tracheomalacia for several years.
Complications
Potential complications include recurrent laryngeal nerve injury, mediastinitis, respiratory distress, and dysphagia. Mediastinitis must be aggressively treated with IV antibiotics. Positive pressure ventilation and pulmonary care are necessary for respiratory distress, although aggressive positive pressure ventilation may compromise the anastomosis in the airway. Dysphagia is a chronic problem; many patients require feeding tube. Input from a skilled pediatric swallow therapist is necessary. Mortality associated with a type 4 cleft approaches 90%.
Outcome and Follow-Up
Sedation and paralysis is employed in the early recovery period. Splinting is useful to maintain the neck in a neutral midline position. An oral or naso-gastric tube is contraindicated; pressure compromises the anastomosis and may result in breakdown, tissue necrosis, and fistula formation. Antireflux medications and good pulmonary toilet are maintained. Enteral feeding is preferable (via jejunostomy or gastrostomy).
Outcomes vary greatly with the extent of anomaly, specific treatment, and possible comorbidities. For type 1 clefts repaired endoscopically, there is a 94% success rate reported in one series.
ICD-9 Code
748.3 | Laryngeal cleft |
Further Reading
Chien W, Ashland J, Haver K, Hardy SC, Curren P, Hartnick CJ. Type 1 laryngeal cleft: establishing a functional diagnostic and management algorithm. Int J Pediatr Otorhinolaryngol 2006;70(12):2073–2079
Chitkara AE, Tadros M, Kim HJ, Harley EH. Complete laryngotracheoesophageal cleft: complicated management issues. Laryngoscope 2003;113(8):1314–1320
Rahbar R, Rouillon I, Roger G, et al. The presentation and management of laryngeal cleft: a 10-year experience. Arch Otolaryngol Head Neck Surg 2006;132(12):1335–1341
6.5 Tracheoesophageal Fistula and Esophageal Atresia
Key Features
• Tracheoesophageal fistula (TEF) and esophageal atresia (EA) are the result of a congenital communication between the trachea and esophagus.
• EA is also present in most cases.
• These congenital anomalies present with respiratory and/or feeding difficulties in the newborn.
Congenital TEF and EA are common congenital anomalies that usually occur together. Most cases are diagnosed immediately following birth or during infancy due to the associated life-threatening complications. However, isolated TEF may escape diagnosis until later. Problems with growth and feeding, pulmonary complications, and gastroesophageal morbidity may result from the condition.
Epidemiology
TEF and EA in their various forms occur in ~1 in 3000 live births, with a slight male predominance. In over 50% of cases, TEF and EA are linked with other defects, such as in the VACTERL sequence and with chromosomal anomalies.
Clinical
Signs and Symptoms
A sign of the condition, though nonspecific, is polyhydramnios on prenatal ultrasound. Symptoms of TEF include recurrent chest infections, cyanosis and choking on feeding, and abdominal distension. When EA is also present, the patient will not be able to swallow saliva or feed, and will drool excessively.
Differential Diagnosis
• Aspiration pneumonia
• Laryngeal cleft
• Tracheomalacia
• Esophageal stricture
• Esophageal diverticula
• Vascular ring
• Gastroesophageal reflux
• Other feeding problems
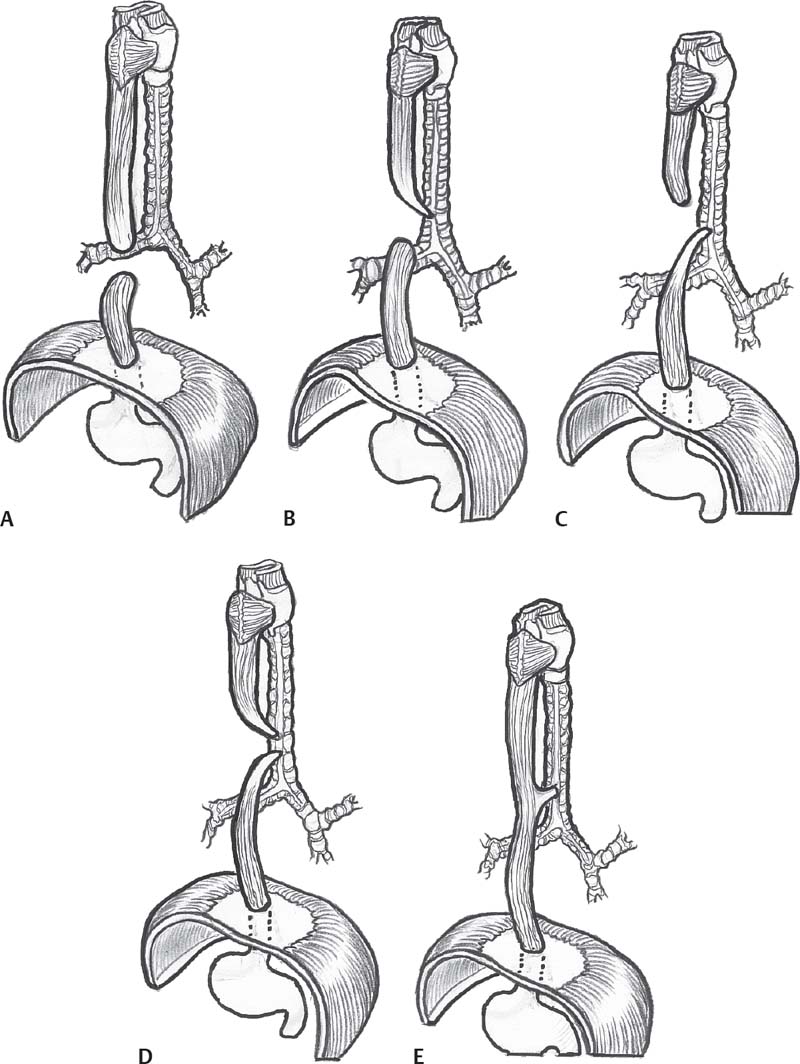
TEF and EA develop as a result of incomplete separation of the respiratory and digestive divisions of the primitive foregut. Several variants of TEF and EA have been described, but five types predominate, as shown in Fig. 6.2.
Evaluation
Physical Exam
The exam is likely to be normal unless marked abdominal distension is present. Other congenital anomalies may be noted. Passage of a tube from the mouth to the stomach may be impossible in the presence of EA. The subsequent chest x-ray will typically show the tip of the catheter coiled in the proximal esophageal pouch.
Imaging
A chest x-ray will show gaseous distension of the bowel when TEF is present. There may be pulmonary changes from persistent respiratory infections. A contrast esophagram can be used to demonstrate an isolated TEF but may provide a false-negative because of the slanted orientation of the fistulous tract. Abdominal ultrasonography is used to look for associated renal pathology. Echocardiography is used to examine the heart for any associated anomaly and to aid in planning surgical treatment. Bronchoscopy may help find the location of the fistula and guide operative strategy.
Treatment Options
To prevent aspiration and gastric reflux, a sump catheter should be immediately placed into the upper esophageal pouch and connected to constant suction. The patient should be placed in a prone, head-up position.
Most infants undergo immediate primary repair. Reasons for the delay of surgical treatment include severe associated anomalies, severe pneumonia or respiratory distress, and a long gap between the esophageal pouches.
Repair consists of ligation of the fistula and primary esophageal anastomosis via a right posterolateral thoracotomy. If a right-sided aortic arch is present, a left thoracotomy is used. A cervical incision may be used if there is an isolated TEF. If more than two vertebral bodies separate the upper and lower esophageal segments (“long-gap”), extramucosal circular myotomies can be used. If the gap persists or if there are significant complications, replacement of the esophagus is necessary.
If a patient cannot undergo immediate primary repair, a gastrostomy may be used for gastric decompression and access for feedings.
Complications
Anastomotic Leaks
Anastomotic leaks usually resolve with parenteral nutrition and posterior drainage. Repeat thoracotomy is required if healing does not occur.
Stricture
Stricture is usually treated with repeated dilatation.
Dysphagia
Peristalsis is abnormal in the vast majority of patients with a history of TEF or EA. Patients are advised to eat slowly and may need to avoid meats. Esophageal obstruction may occur and foreign body removal may be required.
Acid-Reflux Disorders
All patients should be placed on antireflux medical treatments and these should be continued until at least the time when an upright posture is achieved. Antireflux surgery may be necessary in some cases.
Tracheomalacia
Treatment is generally reserved for those with “dying spells” or recurrent pneumonia. Aortopexy is generally employed. If this fails, tracheotomy may be required. Rarely tracheal stents are employed, but they are not used in infants and are uncommonly employed in young children.
Recurrent Tracheoesophageal Fistula
Recurrent TEF requires reoperation.
Outcome and Follow-Up
Oral feeding is delayed until a contrast study done several days after the operation shows no leaks or narrowing around the anastomosis. In the case of isolated TEF repair, oral feeding can resume immediately if the integrity of the repair is certain. Complications occur relatively frequently following operative repair of TEF/EA and one or more additional operations are needed in half the cases. Strictures are the most common complication, and a barium swallow or esophagoscopy is needed before hospital discharge in all patients.
Although complications occur relatively frequently, patients have an excellent chance of leading normal lives in the absence of severe associated anomalies. Inherent structural and functional defects in the trachea and esophagus result in significant respiratory and gastroesophageal sequelae, including poor growth, feeding problems, tracheomalacia, bronchomalacia, recurrent chest infections, and reflux. However, the frequency of such events appears to decrease significantly with age.
ICD-9 Code
750.3 | Tracheoesophageal fistula, esophageal atresia, and stenosis |
Further Reading
Crabbe DCG. Isolated tracheo-oesophageal fistula. Paediatr Respir Rev 2003;4(1):74–78
Goyal A, Jones MO, Couriel JM, Losty PD. Oesophageal atresia and tracheo-oesophageal fistula. Arch Dis Child Fetal Neonatal Ed 2006;91(5):F381–F384
Kovesi T, Rubin S. Long-term complications of congenital esophageal atresia and/or tracheoesophageal fistula. Chest 2004;126(3):915–925
Morrow SE, Nakayama DK. Congenital malformations of the esophagus. In: Bluestone CD, Stool SE, Alper CM et al, eds. Pediatric Otolaryngology. 4th ed. Philadelphia, PA: WB Saunders; 2001
6.6 Vascular Rings
Key Features
• The trachea and esophagus are completely or incompletely surrounded by vascular structures.
• Compression of the trachea, the bronchi, and/or the esophagus may occur.
• Most symptomatic malformations present during infancy or early childhood.
The term vascular ring refers to an aortic arch abnormality in which the trachea and esophagus are surrounded by vascular structures. It may be complete or incomplete. The greater the degree of compression the vascular ring causes, the more severe the symptoms are and the earlier they present. For symptomatic patients, treatment is generally surgical.
Embryology and Anatomy
Vascular rings arise during embryonic development from the abnormal evolution of the arterial branchial arch system. In normal embryonic vascular development, ventral and dorsal aortae are connected by six pairs of aortic arches. The first, second, and fifth arches regress, as well as a portion of the right fourth arch. This leaves the usual left aortic arch. Residual segments of the third, fourth, and sixth arches develop into the mature anatomy of the mediastinal vascular structures. Inappropriate persistence or development of segments leads to congenital aortic arch anomalies.
The most common vascular ring is a double aortic arch, accounting for 50 to 60% of symptomatic vascular rings. A right aortic arch with an aberrant left subclavian artery is the second most common, accounting for 12 to 25% of cases. Other vascular anomalies include a right aortic arch with mirror image branching and left ductus arteriosus, a pulmonary artery sling, an anomalous innominate artery, and left aortic arch anomalies.
Epidemiology
At autopsy, 3% of the population has a congenital anomaly of the aortic arch system. Most are asymptomatic. Vascular rings account for less than 1% of congenital cardiovascular malformations.
Clinical
Signs and Symptoms
Symptoms depend on the location and degree of vascular compression. Wheezing, stridor, aspiration, cyanotic or apneic attacks, and dysphagia are characteristic. Feeding may exacerbate stridor. Dysphagia is worsened by solid foods. Recurrent respiratory infections such as aspiration pneumonia may also be present.
Differential Diagnosis
• Asthma
• Tracheomalacia
• Bronchiolitis
• Laryngeal stenosis
• Congenital stridor
• Laryngeal web
• Croup
• Foreign body aspiration
• Laryngomalacia
Evaluation
Physical Exam
Physical findings vary, often in accordance with the patient’s history. Stridor is characteristically expiratory. It is often associated with cough, tachypnea, and rhonchi. Expiratory, high-pitched wheezes and intercostal retractions can also be appreciated. Patients may hold their neck in hyper-extension to alleviate respiratory distress. Respiratory findings typically do not improve with nebulized bronchodilator treatment and are worsened by exertion. Pulmonary infection may be the presenting symptom, especially in older children.
Imaging
A chest radiograph will reveal laterality of the aortic arch by typical contralateral deviation of the trachea. Two arches may be suspected if there is compression of the trachea at the level of the arches. On the lateral view, anterior tracheal compression may be evident. Tracheal constriction evidenced by narrowing or obliteration of the distal tracheal air column and lung hyperinflation may also be seen.
Bronchoscopy is often the diagnostic technique of choice to evaluate structural and dynamic anomalies of the airway. Bronchoscopy can also evaluate the tracheobronchial tree for coexisting or intrinsic lesions such as tracheomalacia, stenosis, complete tracheal rings or aberrant bronchi. In many centers, MRI is becoming the diagnostic technique of choice. Sedation, airway management, and intubation may be challenging in the patient with signs and symptoms of airway compression.
CT may show location, degree, and extent of tracheal narrowing. It is faster and requires less sedation than MRI but is not as good at defining vascular anatomy unless CT angiography is obtained. Other disadvantages include exposure to ionizing radiation and IV contrast.
Frequently but not invariably, echocardiography can show the presence and define the anatomy of a vascular ring. It will show associated cardiovascular anomalies and can be done at the bedside. It does not image the airways.
Cardiac catheterization with angiography provides a clear delineation of abnormal vessels and is helpful in evaluating associated congenital heart defects. With the advent of MRI, it is rarely necessary for isolated aortic arch anomalies.
Other Tests
Pulmonary function tests can be used in the evaluation of infants and children with suspected tracheal obstruction of vascular origin. The shape of partial expiratory flow-volume curve can help to localize the site and assess the severity of airway obstruction. Usually, pulmonary function testing cannot be obtained in children under age 6.
Treatment Options
Medical
Asymptomatic or mildly symptomatic patients can be managed medically with humidification of inspired air, drainage of bronchial secretions, antibiotics, and supplemental oxygen when needed, and a soft diet or tube feedings if dysphagia exists.
Surgical
Surgery is indicated in all patients with symptomatic vascular rings. Asymptomatic complete rings should undergo elective surgery if there is suspicion for progressive airway compromise. The goal of surgical treatment is to divide the compressive vascular ring, relieve tracheobronchial and esophageal compression, and maintain normal perfusion of the aortic arch. For a double aortic arch, the atretic or hypoplastic arch is divided along with the ligamentum or ductus arteriosus. For right aortic arch variants, the left ductus or ligamentum arteriosum is divided. If surgery is indicated for an anomalous innominate artery, the preferred treatment is aortopexy. For a pulmonary artery sling, the ductus or ligamentum arteriosum is divided and the left pulmonary artery is divided and reimplanted into the main pulmonary artery.
The approach is generally through a left posterolateral thoracotomy, but certain types of vascular rings require a right thoracotomy. A sternotomy incision is indicated for concomitant repair of intracardiac defects, or for the repair of pulmonary artery sling, or for repair of complete tracheal rings with slide tracheoplasty on cardiac bypass. Intraoperative bronchoscopy is helpful in evaluating the effects of surgery on airway patency; tracheomalacia will persist after repair but will usually improve with time.
Complications
Tracheomalacia and Bronchomalacia
Prolonged intubation may be required to maintain airway patency in long-segment malacia but can lead to endoluminal airway complications such as granuloma formation. Endoluminal stenting procedures have also been described. Occasionally, reconstruction of the affected airway segment is required. Generally, with growth of the trachea and gradually increasing stiffness of the cartilage, symptoms are likely to improve. Tracheotomy may be needed to stent substantial tracheomalacia.
Recurrent Laryngeal Nerve Injury
In unilateral vocal fold paralysis, the need for intervention is based on the degree of hoarseness and the risk of aspiration. In bilateral fold paralysis, surgery is needed to alleviate glottic obstruction. Tracheotomy may also be required.
Chylothorax
This is an uncommon perioperative complication. Treatment of choice is implantation of a pleuroperitoneal shunt.
Outcome and Follow-Up
Intensive respiratory care is always needed postoperatively. Humidified oxygen, antibiotic prophylaxis for pulmonary infections, frequent suctioning of tracheal secretions, and diligent chest physiotherapy are vital.
Major postoperative issues relate to concurrent cardiac defects and residual airway disorders. Successful tracheal extubation is possible in most patients. Successful repair of the vascular ring may not immediately relieve airway obstruction.
The patients who are asymptomatic or mildly symptomatic with incomplete rings are likely to improve with age. Of the patients who received surgical repair, 95% are expected to survive, and most of them will become completely asymptomatic. However, persistence of various degrees and types of pulmonary function anomalies may be found in a significant number of patients.
ICD-9 Code
747.21 | Vascular ring, other anomalies of aortic arch |
Further Reading
Bennet EC, Holinger LD. Congenital malformations of the trachea and bronchi. In Bluestone CD, Stool SE, Alper CM, et al, eds. Pediatric Otolaryngology. 4th ed. Philadelphia, PA: WB Saunders; 2001
Kussman BD, Geva T, McGowan FX. Cardiovascular causes of airway compression. Paediatr Anaesth 2004;14(1):60–74
Valletta EA, Pregarz M, Bergamo-Andreis IA, Boner AL. Tracheoesophageal compression due to congenital vascular anomalies (vascular rings). Pediatr Pulmonol 1997;24(2):93–105
6.7 Subglottic Stenosis
Key Features
• The subglottis is the narrowest part of the infant airway.
• Acquired subglottic stenosis is most commonly found; the most common related factor is intubation.
• Subglottic stenosis of 70% of the lumen or greater is associated with daily symptoms.
The subglottis, the narrowest part of the pediatric airway, is composed of a complete cartilaginous ring, the cricoid, and loose submucosa that swells when irritated (as in, for example, croup). It is the region most likely to be affected by pressure from a too-large or frequently moving endotracheal tube. Congenital subglottic stenosis most commonly results from an abnormally shaped cricoid, with intraluminal lateral shelves, resulting in an oval shape to the lumen. Intervention is individualized to the patient: sometimes a watch and wait approach works; other children benefit from some type of surgery.
Epidemiology
Incidence is ~1.5 cases per million, but it occurs in 1 to 8% of neonates requiring intubation.
Clinical
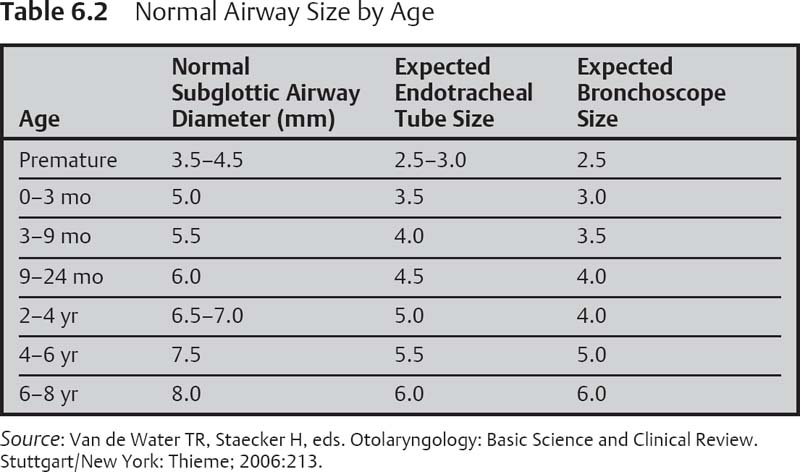
An abnormal, narrow airway causes stridor. It is, therefore, worth reviewing the typical dimensions of the “normal” airway (Table 6.2).
Signs
The signs include stridor, respiratory distress, and typically a normal voice.
Symptoms
• Unsuccessful extubation of a patient in the neonatal intensive care unit (NICU)
• Stridor, may be biphasic, may present only with agitation
• Respiratory distress exacerbated by upper respiratory tract infection
• Recurrent croup
• Feeding problems
• Slow growth, failure to thrive
Differential Diagnosis
Other causes of stridor with normal voice include laryngomalacia, subglottic cyst, subglottic hemangioma, and tracheal stenosis.
Evaluation
Physical Exam
Flexible laryngoscopy should be done to assess vocal fold movement. The subglottis is sometimes seen with this exam, but the scope should not be passed through the glottis because of the risk of inducing vasovagal reflexes. Bronchoscopy will show subglottic stenosis. The degree of narrowing and the length of the narrowed segment are important to measure.
The Cotton-Meyer grading system apparently correlates to symptoms and prognosis (Table 6.3). Grade I stenoses typically are asymptomatic unless there is an upper respiratory tract infection.
Grade | Degree of Narrowing |
|---|---|
I | 50% |
II | 50–70% |
III | 71–99% |
IV | Total obstruction |
Imaging
A soft tissue lateral x-ray, airway fluoroscopy, or CT may show the subglottic anatomy. Bear in mind that airway fluoroscopy involves significant radiation exposure.
Other Tests
A complete evaluation for reflux (including a barium swallow, a gastric scintiscan, a pH probe) may be helpful. Pulmonary function tests and video-stroboscopy are indicated in cooperative older patients.
Pathology
Congenital subglottic stenosis may be cartilaginous or membranous (a thickened submucosa). The subglottis is the narrowest part of the pediatric airway in normal circumstances, so pressure necrosis from an endotracheal tube is more likely here than elsewhere in the airway. Acquired stenosis may be caused by mucosal necrosis with healing by granulation tissue and subsequent fibrosis, although deeper injuries including cartilage necrosis can occur. Factors related to injury include size of endotracheal tube, number of reintubations, tube movement, and tube material (polyvinyl chloride is considered safest).
Treatment Options
Medical
Antireflux medications have a role in management. If an acid-reflux disorder is not diagnosed with traditional tests, prophylactic reflux medications should be given, especially if there is a suggestion of reflux laryngitis on examination.
Surgical
Dilation
Dilation is an option for mild soft stenosis.
Laser Division
Laser division (CO2, argon, or KTP laser) is an option for early stenosis (granulation tissue), crescent-shaped bands, and thin circumferential webs.
Cricoid Split
Laryngotracheoplasty
If more than 3 mm of circumference gain is needed, a cartilage graft is necessary. Auricular or costal cartilage may be used. Grafts can be placed anterior and posterior in the subglottis. Stenting is required if the posterior cricoid is divided; this may be achieved by endotracheal intubation for 1 to 2 weeks.
Only mature stenoses should be reconstructed with an open procedure. Cricotracheal resection is reserved for amenable airway lesions.
Complications
Emergency complications can involve airway obstruction or respiratory problems. Causes include mucous plugs, granulation tissue, aspiration of stenting materials (if used), hematoma, hemorrhage into the airway, or pneumothorax. Treatment is aimed at the underlying problem. Treatment may require airway suctioning for mucous plugs, use of aerosolized steroids (e.g., dexamethasone 0.25 to 1.0 mg/kg per day, to a maximum of 20 mg/d) to reduce granulations, chest tube placement for pneumothorax, hemorrhage or hematoma, or return to the operating room for bronchoscopic or possible open treatments.
Other complications include cricoid split failure (repeat intubation for 72 additional hours with a half-size smaller tube; if that fails, then tracheotomy) and laryngotracheoplasty failure (infection may lead to graft necrosis, treated by antibiotics and revision surgery).
Outcome and Follow-Up
A postoperative chest radiograph should always be obtained for open reconstructive procedures. Continuous O2 saturation monitoring should be overnight; an ICU setting should be available for any open procedures. Sedation is required but paralysis is not desired, so that in the event of accidental extubation, the child can make breathing efforts. Also, spontaneous ventilation is preferred because of improved airway clearance of secretions and decreased muscle weakness that can develop from not breathing for 7–10 days.
Antireflux medications are typically employed. Prophylactic postoperative antibiotics are typically given. Management by a skilled pediatric intensivist is required, to reduce potential morbidities.
These children require long-term follow-up. Depending on the procedure employed and the patient’s symptoms, serial bronchoscopy may be indicated.
ICD-9 Code
478 | Other diseases of upper respiratory tract |
Further Reading
Cotton RT, Andrews TM. Laryngeal stenosis. In: Bailey BJ, Johnson JT, Newlands SD et al, eds. Head and Neck Surgery – Otolaryngology. 2nd ed. Philadelphia, PA: Lippincott-Raven; 1998:1115–1130
Greinwald JH, Cotton RT. Pathophysiology of stridor and airway disease. In: Van de Water TR, Staecker H, eds. Otolaryngology: Basic Science and Clinical Review. Stuttgart/New York: Thieme; 2006:212–224
McMurray JS, Myer CM. Management of chronic airway obstruction. In: Wetmore RF, Muntz HR, McGill TJ, et al, eds. Pediatric Otolaryngology: Principles and Practice Pathways. Stuttgart/New York: Thieme; 2000:863–882
Myer CM, Cotton RT, Shott SR. The Pediatric Airway: An Interdisciplinary Approach. Philadelphia, PA: JB Lippincott; 1995
6.8 Pierre Robin Sequence
Key Features
• Pierre Robin sequence requires the presence of micrognathia, glossoptosis, and usually a cleft palate (frequently U-shaped).
• Infants present with airway obstruction because the tongue falls into the pharyngeal airway, and feeding difficulties.
• Hearing loss is frequent, from otitis media (OM) with effusion (OME; 90%), middle ear anomalies (60%), and inner ear anomalies (40%).
First described in 1891 by Lannelongue and Ménard, then further by Pierre Robin in 1923, the sequence requires the presence of micrognathia and glossoptosis. Most (90%) also have a cleft palate. Neonates have feeding difficulties. They have upper airway obstruction and are usually difficult to intubate. These children have a high incidence (up to 80%) of other systemic anomalies.
Epidemiology
Clinical
Signs
• Micrognathia, glossoptosis, cleft palate
• Airway obstruction with desaturations
• Pinna abnormalities, OME may be associated
Symptoms
• Airway obstruction with stertor, cyanosis, respiratory failure
• Prone position may improve airway obstruction in mild cases
• Failure to thrive
Differential Diagnosis
• Stickler syndrome
• Velocardiofacial syndrome
• Fetal alcohol syndrome
• Treacher-Collins syndrome
• Nager syndrome
• Beckwith-Wiedemann syndrome
All of these syndromes are associated with Pierre Robin sequence.
Evaluation
The most important issue is to first ensure an adequate airway and feeding. Then, one should determine whether there is an associated syndrome. Syndromic patients will generally require more involved interventions.
Physical Exam
A complete head and neck exam will reveal the signs of the sequence. A maxillary–mandibular discrepancy can be measured by placing the infant upright, passively closing the jaw, placing the wooden end of a cotton applicator on the anterior surface of the mandibular alveolar ridge in the midline, then marking where the anterior surface of the maxillary alveolar ridge falls. This measure can be used to monitor growth and surgical outcome.
Pinnae and tympanic membranes should be evaluated. A flexible laryngoscopy is required to rule out concomitant laryngomalacia.
Imaging
Imaging is not typically required to assess the airway obstruction. Imaging may be indicated to evaluate other anomalies coincident with the sequence.
Labs
Continuous monitoring and oxygen saturation monitoring in a NICU environment is required.
Other Tests
All children with Pierre Robin sequence should have early vision and hearing screening, given the association with Stickler syndrome. A sleep study may be indicated in mild cases. Bronchoscopy is indicated in severe cases.
Pathology
This sequence is initiated by mandibular hypoplasia in utero. Because of insufficient room in the mouth for the tongue, it remains positioned between the palatal shelves preventing their normal fusion and leading to a cleft palate.
Treatment Options (Table 6.4)
Medical
• Positioning: prone position works in about half of patients
• Nasopharyngeal airway
• Oral airway
• Short term intubation
• Manage reflux
Prone positioning |
Nasopharyngeal airway |
Glossopexy, tongue–lip adhesion |
Tracheotomy for severe obstruction, synchronous airway lesions, failure of other methods |
Mandibular distraction osteogenesis |
Surgical
About half of patients require surgery to support their airway.
Temporary Tongue–Lip Adhesion
This involves raising a flap on the inner lower lip and ventral tongue, suturing these together, and sometimes suspending a button placed on the tongue base via a suture that goes around the anterior mandible (this button stays in only for the first week). This brings the tongue forward. It is all taken down by the first year of life; by this time the mandible has usually grown enough that the airway is clear. Fifteen percent will fail and require a tracheotomy.
Mandibular Distraction Osteogenesis
Proximal and distal screws are placed into the mandible bilaterally, then an osteotomy is performed between them. An external device is attached to gradually distract the mandibular segments, typically 1 mm per day. The distractor needs to be left in place for 6 to 8 weeks for consolidation.
Tracheotomy
A tracheotomy is indicated in syndromic children, patients with aspiration, patients with reflux or severe sleep apnea, those with second sites of obstruction below the hypopharynx, and those who fail tongue–lip adhesion and/or distraction osteogenesis.
Complications
• Tongue–lip adhesion: Dehiscence or failure to relieve airway obstruction is treated by either mandibular distraction or tracheotomy.
• Mandibular distraction osteogenesis: Pin tracks may become infected, loosening the pins, which will then require replacement; note also that tooth buds may be damaged. TMJ ankylosis and malocclusion problems may occur.
• Tracheotomy (see A6 in Appendix A)
Outcome and Follow-Up
Infants require continuous close monitoring in an ICU setting. Initial feeding requires a nasogastric tube, and if there are no desaturations, trials of oral feeding start. Children may be discharged home once the airway and feeding are stabilized.
Elective repair of the cleft palate is also required, but management of airway obstruction takes priority. Nonsyndromic patients can generally undergo tracheotomy decannulation following palatoplasty, if standard decannulation criteria are met.
Nonsyndromic children, particularly those not requiring surgical intervention, do very well with catch up growth and are likely to have a normal facial profile by age 5. Syndromic children are more likely to require multimodality treatment. Follow-up only needs to continue until the airway obstruction is resolved.
ICD-9 Codes
749 | Cleft palate |
748 |



