From Gomez Morales A. Coats’ disease. Natural history and results of treatment. Am J Ophthalmol 1965;60:855–864; Shields JA, Shields CL, Honavar SG, et al. Classification and management of Coats’ disease: the 2000 Proctor lecture. Am J Ophthalmol 2001;131:572–583.
EPIDEMIOLOGY (PREVALENCE, ENVIRONMENTAL FACTORS)
The epidemiologic and clinical features described in recent clinical series correspond with that originally reported by Coats. Cases can present as early as the first month and as late as the eighth decade of life, but median age at presentation is 5 years (18), and approximately two-thirds of patients present before age 10 years (14,15,18,19). Younger patients appear to be afflicted more rapidly and with more severe progression. In a series of 75 patients with advanced disease reported by Haik (20), 30 (40%) presented before age 2 years. At all ages, the disease usually is unilateral (90%); if bilateral, it shows asynchronous progression (10,18,21,22). From 70% to 90% of affected children are boys. No racial or ethnic predilection has been shown. Though original demographic studies were performed in Western nations, the male preponderance and the more severe presentation in younger patients are observed in Asian populations as well (23). The precise incidence and prevalence of Coats disease are unknown. No environmental factors have been shown to cause or influence the severity of Coats disease.
A genetic cause for Coats disease has been proposed based on several observations: (i) the association of retinal telangiectasias with muscular dystrophy and deafness in one family (24), (ii) occurrence of exudative retinopathy in a few members of families with retinitis pigmentosa (25–27), and (iii) the case of a mother purported to have unilateral retinal telangiectasias and a son with Norrie disease (28). Cytogenetic studies of isolated cases have demonstrated pericentric inversion of chromosome 3 in one child and a partial deletion of chromosome 13 in another (29,30). However, the overwhelmingly sporadic occurrence of Coats disease precludes any substantive genetic linkage studies.
Black et al. (28) investigated a mother with “a unilateral variant of Coats disease” and her son afflicted with Norrie disease. Both carried a missense mutation in the Norrie disease (NDP) gene, which has been implicated in retinal vasculogenesis (31). Archived tissue from nine eyes enucleated for Coats disease then was analyzed, and a mutation in the NDP gene was found in retinal tissue from one eye. The authors postulated that Coats telangiectasias arise from somatic mutation in the NDP gene (28). The possibility that the mutation represented a naturally occurring polymorphism was not conclusively eliminated, however.
In a study of five families with a specific form of retinitis pigmentosa (designated RP12), five of eight patients with an associated Coats-like exudative vasculopathy demonstrated mutations in the CRB1 (crumbs homolog 1) gene. It remains unclear, however, whether the exudative changes seen in such cases are truly an independent genetic event or merely are secondary to vascular endothelial decompensation associated with the retinal degeneration. Mutations in the CRB1 gene also are seen in patients with RP12 and a small percentage of patients with Leber congenital amaurosis (32). The CRB1 gene has been examined in patients with primary, unilateral Coats disease without associated RP. Mutation analysis of the CRB1 gene in 18 unrelated patients with primary, unilateral Coats disease revealed no variants in the gene (33).
Coats disease is a rare disorder and does not impact many children throughout the world; however, the disease can have a negative impact on the quality of life of those who are affected. Coats disease is included on the National Organization of Rare Disorders Web site (http://www.rarediseases.org).
The cause of Coats disease remains unclear. Reese (5) observed periodic acid-Schiff staining of basement membrane under the endothelium of retinal veins and theorized that deposition of polysaccharide led to atresia and occlusion of vessel lumina, “thereby occasioning vascular ectasia and the formation of collateral channels.” Wise (34) speculated that local retinal hypoxia awakened “a dormant vasoproliferative factor,” stimulating growth of new vessels from veins and capillaries. Elevated levels of such a factor, namely, vascular endothelial growth factor (VEGF), have been documented in the aqueous and subretinal fluid of eyes with advanced Coats disease (35).
Histologic and ultrastructural studies support Coats’ original speculation “that some of the vascular changes are primary” and lead to the classic histopathologic findings of vessel thickening and hyalinization, interspersed with thinning and loss of endothelial elements (1,22,36). It is believed that breakdown of the blood–retinal barrier, at the level of the endothelium, causes plasma leakage into vessel walls, which become necrotic and disorganized and form dilatations and telangiectasias. Further leakage into adjacent retinal tissue produces the recognizable intraretinal and subretinal cholesterol exudates, hemorrhage, cysts, edema, lymphocytic infiltration, and deposition of lipid and fibrin (37,38). These changes lead to degeneration of the neural retina and to infiltration by phagocytic, lipid-laden “ghost” cells, which appear to be transformed retinal pigment epithelial cells (39). Clinically, these microscopic changes manifest as irregular, raised patches of yellow-white or yellow-green material, often with superficial hemorrhage. Partial serous retinal detachment may ensue and worsen with increasing vasculopathy and exudation. The exudation can be so abundant that it drains into the orbit and stimulates an inflammatory reaction (33).
CLINICAL FEATURES, SYMPTOMS, AND SIGNS
The most common signs of disease are strabismus, leukocoria, and visual impairment detected on routine vision screening. One child presented with turbid yellow fluid filling the anterior chamber (40). Up to 25% of patients can be asymptomatic, with diagnosis occurring during routine ophthalmic examination (41). The hallmark funduscopic findings in Coats disease are vascular telangiectasias and massive subretinal and intraretinal exudates (Fig. 52.1). Affected vessels display an irregular caliber, focal telangiectasias, aneurysmal dilatations (“light bulbs”), and sheathing by yellow cholesterol deposits. Microaneurysms can occur in all parts of the vascular bed but most commonly arise from capillaries (6). In the series of Henkind and Morgan (42), vascular abnormalities, although not always apparent clinically, were found histologically in all cases.
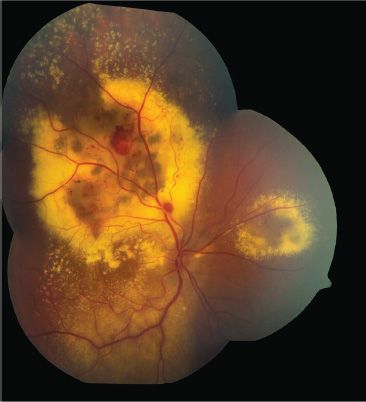
FIGURE 52.1 Montage fundus photograph of the right eye of a 10-year-old boy with retinal vascular abnormalities and exudation characteristic of Coats disease.
The earliest changes involve the equatorial and peripheral retina, with the temporal quadrants and the sector temporal to the fovea most commonly involved (19,43). The posterior pole is involved less frequently than the periphery (6,19,20,44). In a series of 112 eyes, Spitznas et al. (6) found lipid deposits in the central retina in fewer than 50%, with concomitant vascular changes in 17%. Shields et al. (18) observed retinal telangiectasias restricted to the macula in 1%.
The macula can become involved directly, by exudation from macular telangiectasias or indirectly by accumulation of exudate from peripheral retinal telangiectasias (Figs. 52.2 and 52.3) (4). The macular exudate is yellow and occurs in continuous broad sheets that can form subretinal mounds (10,16). Macular edema and exudative macular detachment can ensue, resulting in significant visual decline (4). Advanced macular disease can be associated with macular fibrosis and formation of subfoveal nodules (Fig. 52.3) and macular holes, possibly secondary to abnormalities of the vitreomacular interface (45,46).
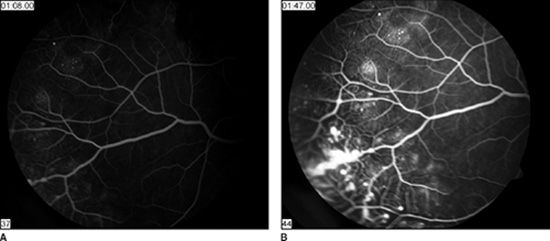
FIGURE 52.2 Fluorescein angiography of the retinal periphery of the same eye shown in Figure 52.1. A: The early phase of the angiogram shows typical vascular changes of capillary dilatation, retinal nonperfusion, telangiectasias, and microaneurysms. B: The late phase of the angiogram shows leakage arising from microaneurysms (so-called light bulbs) and from generalized vascular hyperpermeability.

FIGURE 52.3 Wide-field fundus photograph of the right eye of an 8-year-old boy with dense macular exudation and only retinal vascular changes in the inferotemporal periphery. Over time, some of the macular exudates have coalesced to form a subfoveal nodule.
The optic disc can appear hyperemic (10). The vitreous remains clear until advanced stages. Then, vitreous condensation and contraction of vitreoretinal connections lead to retinal detachment and vitreous hemorrhage. Intraretinal macrocysts can develop, likely as a result of long-standing retinal detachment (18,44).
In the early stage of Coats disease, ocular abnormalities outside the fundus are rare but many of the telangiectasia are located in the peripheral retina. Isolated associated findings include congenital retinoschisis, uveal coloboma, choroidal angioma, morning glory disc, and microphthalmos (13,20,41,47,48). There is one report of bilateral Coats disease associated with infantile cataracts, congenital glaucoma, and later ketotic hypoglycemia (49). A fundus appearance resembling Coats disease is seen in up to 4% of patients with retinitis pigmentosa (50).
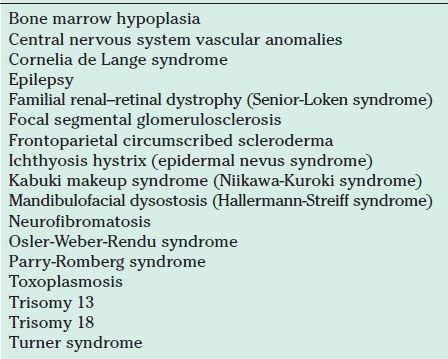
The overwhelming majority of children with Coats disease are otherwise healthy, but Coats disease has been observed rarely in patients with various systemic disorders, as listed in Table 52.2 (51–63). In particular, patients with facioscapulohumeral muscular dystrophy display retinal changes typical of incipient Coats disease (24,64). In a cohort of 64 patients with this disorder, retinal telangiectasias were documented angiographically in 48 (75%) (65). The strong overlap of these two disorders has been a source of inquiry into a possible genetic source of Coats disease.
Intravenous fluorescein angiography is helpful in children with Coats disease, for both diagnosis and identification of treatable areas. Retinal telangiectasias produce the characteristic “light bulb” appearance and fluoresce dramatically. Capillaries can appear dilated but more often are occluded and are replaced by large arteriovenous shunts within areas of nonperfused retina (6,19). In the late phases of the angiogram, fluorescein dye can leak from aneurysmal vessels to produce a pattern of cystoid macular edema or subretinal pooling (13). Studies reporting nonperfused retina in the fellow eye provide support that Coats disease may have a genetic component or is a generalized process (66).
Ultrasonography provides safe and effective evaluation of leukocoria in which the diagnosis of Coats disease is uncertain. Findings supporting the diagnosis of Coats disease include poorly mobile retinal detachment, convolution and looping of the peripheral retina, dispersed subretinal cholesterol opacities exhibiting constant slow convection movements, and absence of a mass lesion or calcification (67). However, there can be overlap of findings for different diagnoses depending on the diseases and their stages.
Computed tomography (CT) facilitates the differentiation of Coats disease from retinoblastoma. Intraocular calcifications are much more common in retinoblastoma and are readily seen radiographically. Partially calcified nodules can occur in Coats disease but almost exclusively in phthisical eyes (28,44,68). More typical CT findings in Coats disease are homogeneous opacification of the vitreous cavity, homogeneous subretinal densities, distinct retinal detachment, and lack of subretinal enhancement following administration of iodinated contrast dye (20,69).
Use of magnetic resonance imaging (MRI) provides greater tissue delineation, thus allowing differentiation between solid intraocular tumors, that is, noncalcific or minimally calcified exophytic retinoblastoma, and nonneoplastic conditions causing retinal detachment. In Coats disease, there is hyperintensity of the subretinal space on T1-weighted images, hyperintensity or hypointensity of the subretinal space on T2-weighted images, and linear enhancement of retinal detachment following infusion of gadolinium contrast dye (70). The variation in MRI findings likely relates to the variability of the composition of subretinal exudate and the extent of retinal detachment (71).
Analysis of subretinal fluid obtained intraoperatively by fine needle aspiration can confirm the diagnosis of Coats disease and permit safe and more thorough drainage. Fine needle aspiration is not recommended as part of routine evaluation because of the risk of seeding the orbit with malignant cells in cases of retinoblastoma.
Accurate diagnosis of Coats disease, especially in advanced stages, can be difficult. In a series of 62 histologically confirmed cases, Coats disease was the primary clinical diagnosis in only 13 (21%) (44). It must be distinguished from other conditions, some of them life threatening, that cause leukocoria, strabismus, or intraocular mass lesions. The differential diagnosis of Coats disease is given in Table 52.3.
TABLE 52.3
Differential diagnosis of juvenile Coats disease
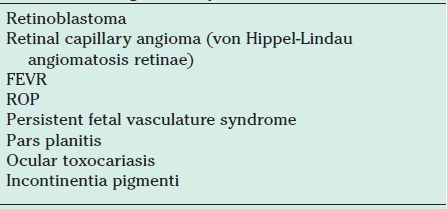
FEVR, familial exudative vitreoretinopathy; ROP, retinopathy of prematurity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


