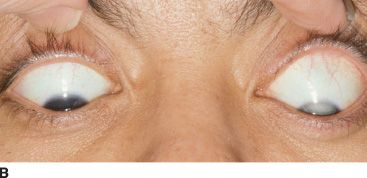
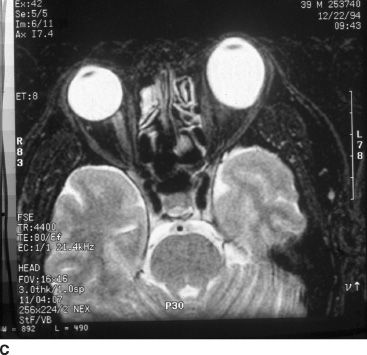
FIGURE 51.1 A: External image of a man with a dense traumatic cataract since early childhood resulting in leukocoria and stimulus deprivation–related increase in axial length of the left eye. B:Note the marked curvature of the lower eyelid during downward gaze due to asymmetry in axial length, mimicking Munson sign as typically seen in patients with keratoconus. C:T2-weighted magnetic resonance image demonstrating marked asymmetry in axial length, with an appreciably larger left eye.
Syndromic Myopia
Ocular—Twin studies suggest a high degree of heritability (~ 85%) (21) of spherical equivalence, raising the question of impact of parental refractive error. Several investigators have described genetic loci associated with nonsyndromic high myopia. Both X-linked (MYP1) (22) and autosomal loci (18p11.31 designated MYP2, 12Q23.1–24 designated MYP3, and 7q36 for AD high myopia) (23–25) have been reported.
Systemic—Genetic loci have been identified for a variety of systemic syndromes having myopia as a component. The most common of these are Stickler syndrome and Marfan syndrome.
The Stickler syndromes (hereditary or progressive oculoarthropathy) type 1 and type 2 occur as a consequence of collagen mutations. Myopia, an “optically empty vitreous,” and retinal detachment are frequent ocular features. The most common form is associated with the 54 exon-containing gene located on chromosome 12q13.11–p13.2 that codes for type II collagen (type 1 Stickler, collagen 2A1) (26). The less commonly encountered form for Stickler syndrome (type 2) is due to a defect in the collagen 11A1 gene involved in type XI collagen production (27). Such patients have a fibrillar or “beaded” vitreous. A mutation in the collagen 11A2 gene results in a systemic Stickler phenotype without ocular findings (28).
Deafness, orofacial features, and arthritis are associated components of type 1 Stickler syndrome. More than 17 mutations have been described for this highly penetrant type 1 gene (29), with considerable phenotypic variability. Some patients present without associated systemic features and with a fundus appearance characterized by a radial perivascular retinal degeneration. This vitreoretinal degeneration may be visible in the first decade, manifesting with retinal holes and tears occurring within the lesion and at its margins (30,31).
Wagner disease (32) is often lumped together with Stickler syndrome due to the shared clinical features of optically empty vitreous and retinal changes. However, the entities are distinct; retinal detachment was not noted in the original description of Wagner disease, and osteoarthropathy is not a feature. Wagner disease has not been demonstrated to exist beyond a single pedigree (33), in which the genetic abnormality has been localized to chromosome 5q13–14 (34). Subsequent examination of 60 members of Wagner’s original pedigree revealed a low incidence of rhegmatogenous retinal detachment, high prevalence of peripheral tractional retinal detachment, and chorioretinal atrophy and cataract in older affected individuals (35). Other vitreoretinopathies have been localized to the same chromosome (36).
Marfan syndrome is due to a mutation in the fibrillin 1 gene (FBN1, locus 15q21.1) (37–39). Skeletal, cardiovascular, and ocular findings are characteristic of the autosomal dominant syndrome. Ectopic lentis with superior temporal displacement, axial high myopia with scleral thinning, and retinal detachment are common ocular findings. Retinal detachment occurs in 5% to 11% of patients, increasing to 8% to 38% in association with ectopic lentis or following lens surgery (40–42). Retinal detachment typically occurs in the third decade, but can be seen in children. There is a high relative prevalence of giant retinal tears and bilateral retinal detachment, total retinal detachment, macula-off retinal detachment, and proliferative vitreoretinopathy, which can commonly present on initial presentation (43).
Additional systemic syndromes with myopia as a component include Knobloch syndrome (collagen 18A1, associated with neuronal migratory defects and mapped to 21q22.3) (44,45) and osteogenesis imperfecta type 1 (collagen1A2) (46), among others (47,48). It should also be recognized that other syndromic collagen defects yet to be named exist, and these can be recognized by a very syneretic vitreous in young individuals. Children with this unusual syneretic vitreous early in life should be managed by more frequent examinations to avoid the circumstance of the initial presentation being that of a large retinal tear and proliferative vitreoretinopathy (PVR) in one eye and symptoms just recognized in the now involved second eye. This situation is common in Marfan syndrome. Therefore, children with syneretic vitreous require more frequent examinations.
Prevention of Myopia in Otherwise Healthy Infants and Children
Over the years, several rationales for the possibility of prevention of myopia have been proposed. The main approach is to prevent a slow myopic progression in otherwise normal children by either pharmacologic (muscarinic antagonists such as atropine or pirenzepine) (49) or refractive means (bifocals in place of single-vision lenses [50] or rigid contact lenses [51,52]). A topical formulation of pirenzepine is under evaluation in a clinical trial (the Myopia Control Study). In general, however, the pharmacologic and refractive strategies have not yielded clinically significant success to date. Most practitioners do not propose these treatments.
Prevention of Nonhereditary Myopia in Infants and Children
A significant opportunity is available to minimize myopia in association with peripheral retinal ablation for severe ROP. The available body of clinical data taken together supports the notion that laser photocoagulation by far is the preferred method of treatment (15).
Infantile vitreous hemorrhage may occur in several clinical scenarios, most commonly in normal infants as a result of birth or nonaccidental trauma or secondary to thrombocytopenia or ROP. Bleeding from a persistent hyaloid artery can also cause a spontaneous vitreous hemorrhage in infants. Although the data are limited, significant stimulus deprivation in a newborn due to dense vitreous hemorrhage has been reported to produce axial myopia on the order of −11 diopters and amblyopia within a matter of 4 weeks (20). Due to the formed nature of the infantile vitreous, waiting in anticipation of spontaneous clearance is not a practical clinical option as clearing is unlikely in the nonprematurely born infant. Lens-sparing vitreous surgery is indicated within 7 to 10 days of onset in such eyes. There are, however, others who suggest that even in unilateral vitreous hemorrhage a wait of up to 6 or even 9 weeks is possible without inducing either axial myopia or a deprivation amblyopia.
Management of Complications of Syndromic Myopia in Infants and Children
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


