FIGURE 43.1 Factors contribute to the development of ROP. Low serum insulin-like growth factor-1 (IGF-1) and its binding protein 3 IGFBP3 derived from preterm birth lead to impaired retinal vascular development. At birth, particularly in the units that have not implemented oxygen monitoring, relatively high oxygen concentration increases ROS generation, which can damage newly formed endothelial cells and leads to capillary constriction. Postnatal oxygen stresses including fluctuations in oxygen delay ongoing physiologic retinal vascular development, leading to the phase of reduced retinal vascularization. Reduced retinal vascularization and increased oxygen demand by photoreceptor differentiation cause retinal hypoxia, which leads to uncontrolled disordered angiogenesis and results in intravitreal neovascularization, the phase of vasoproliferation.
Postnatal Stresses That Increase Risk of ROP
Postnatally, stresses associated with increased risk of ROP include fluctuations in oxygen (5), poor postnatal weight gain (6), perinatal morbidities including sepsis and necrotizing enterocolitis, and other complications of prematurity (7). Several of these can be linked to increased generation of oxidative compounds that can injure blood vessels and/or activate pathways of apoptosis or angiogenesis, and these processes contribute to the biologic features of avascular retina and intravitreal neovascularization in ROP (5).
Postnatal Oxygen: When ROP was first described in the 1940s prior to the implementation of oxygen regulation, very high oxygen likely caused damage to newly formed capillaries and led to ROP (8,9). To study the causes of ROP described in the 1940s, several models of oxygen-induced retinopathy (OIR models) were developed in which animals that vascularize their retinas postnatally were exposed to high levels of oxygen (10–13). The results of this early research led to improvements in oxygen monitoring and the avoidance of excessive, unregulated oxygen at birth (14). However, as infants of younger gestational ages and smaller birth weights survived, ROP reemerged. It is recognized now that, besides high oxygen at birth, postnatal fluctuations in oxygen levels are also associated with severe ROP. It is not clear if it is external fluctuations in inspired oxygen alone that are associated with ROP risk or those caused secondarily from bradycardia, apnea of prematurity, ventilation–perfusion deficits related to lung disease, and other conditions of prematurity. However, healthy newborn animals exposed to repeated fluctuations in inspired oxygen consistently develop features of acute severe ROP supporting causality between oxygen fluctuations and severe ROP features (13). There are also conflicting reports about the role of supplemental oxygen in the development of severe ROP. The Supplemental Therapeutic Oxygen for Prethreshold Retinopathy of Prematurity study found no adverse effect from supplemental oxygen and perhaps a beneficial effect in a subgroup (15). Others have advocated later supplemental oxygen to reduce the risk of severe ROP (16,17). However, several studies have also found correlations between high oxygen saturations and ROP (18,19).
Therefore, the preterm infant at risk of ROP has reduced ability to form normal retinal vessels necessary for the developing retina because of loss of maternal supply of necessary growth factors, high oxygen concentration at birth that can injure newly formed capillaries, and then later events that delay ongoing retinal vascular development (Fig. 43.1). Currently, there is no available technique to accurately measure retinal tissue oxygenation in the human infant in vivo, but experimental evidence provides support that retinal hypoxia exists in temporal association with disordered angio-genesis (20–22). Retinal hypoxia is believed to cause uncontrolled disordered angiogenesis that grows outside the retinal plane and into the vitreous as intravitreal neovascularization, rather than into the retina as postnatal retinal vascularization. Most studies strive to understand the mechanisms leading to this intravitreal neovascularization in ROP and the preceding phase of reduced retinal vascularization. The ultimate goal for ROP is to promote retinal vascularization of the avascular areas and thereby inhibit intravitreal neovascularization, and the later stages 4 and 5 ROP.
Because oxygen has long been associated with ROP, a number of animal models of OIR are used to model acute severe ROP and study how oxygen levels activate signaling pathways that cause features of severe acute ROP. Most models mimic the vascularly active phases of acute severe ROP, that is, plus disease and intravitreal neovascularization (stage 3 ROP), and do not generally develop fibrovascular features, that is, stages 4 and 5 ROP. Currently, stages 4 and 5 ROP are studied in preterm infants undergoing surgery for retinal detachment.
Oxidative Compounds: The preterm infant is predisposed to increased oxidative compounds because of inadequate antioxidant capacity; increased reactive oxygen species (ROS) generated from hypoxia, hyperoxia, and fluctuations in oxygen concentration; and the high metabolic rate and rapid rate of oxygen consumption of the developing photoreceptors (3). The major sources of ROS generation in living cells are the mitochondria and other enzyme systems within the cell that include NADPH oxidase, lipoxygenase, nitric oxide synthetase (NOS), and others. ROS can trigger angiogenic signaling pathways, involving vascular endothelial growth factor (VEGF, referring to VEGFA), and inflammatory signaling pathways via tumor necrosis factor-alpha or other cytokines (21). The retina is particularly vulnerable to ROS because polyunsaturated fatty acids, abundant in the retina, can form oxidative compounds that then cause oxidation in neighboring tissues. Therefore, if excessive ROS are not quenched, tissue damage can result. However, low levels of ROS have physiologic functions to regulate cell growth, survival, differentiation, and metabolism, which all benefit the developing preterm infant. In addition, ROS are also an important defense against infectious agents, critical to the preterm infant with reduced immune defenses. Therefore, a balance in ROS generation and quenching is needed for physiologic growth and to prevent pathologic processes in the preterm infant, and, in concordance with this premise, clinical trials testing antioxidants to prevent ROP have not yielded clear-cut results (23,24).
MODElS OF ROP (OXYGEN-INDUCED RETINOPATHY)
It is virtually impossible to study mechanisms of ROP in human preterm infant’s eyes, because it is unsafe to obtain retinal samples, and biochemical and signaling pathways affected in retina may not be affected in other tissues that are easier to sample. A great deal learned about ROP first came from clinical observations driving research. Subsequently, models were developed to identify genes regulated (25) or proteins activated that caused features of severe ROP. Animals that vascularize their retinas after birth (cat [22], mouse [10], rat, and beagle [11]) were subjected to oxygen stresses and developed features of severe ROP (Table 43.1). These models have limitations. The animals used are not premature; therefore, the effect of oxygen concentration may differ greatly from what occurs in preterm infants. The retina is complex with numerous cells, and interactions between retinal cells can be difficult to study in
TABLE 43.1
Animal models of retinopathy of prematurity (oxygen-induced retinopathy)
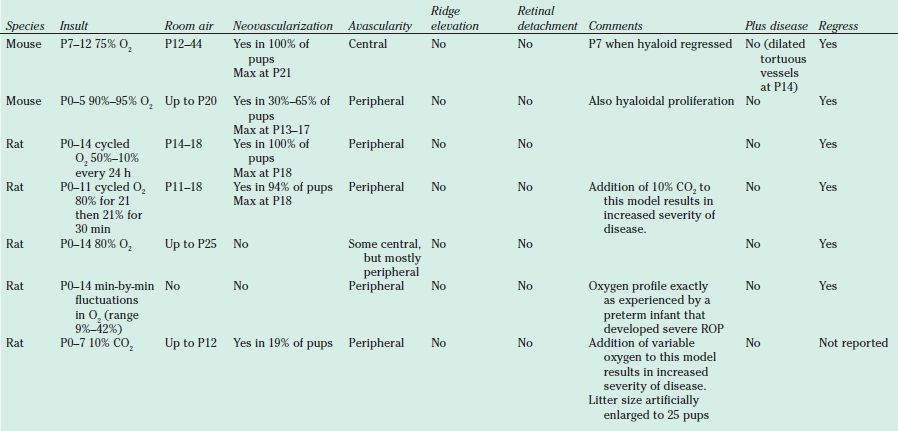
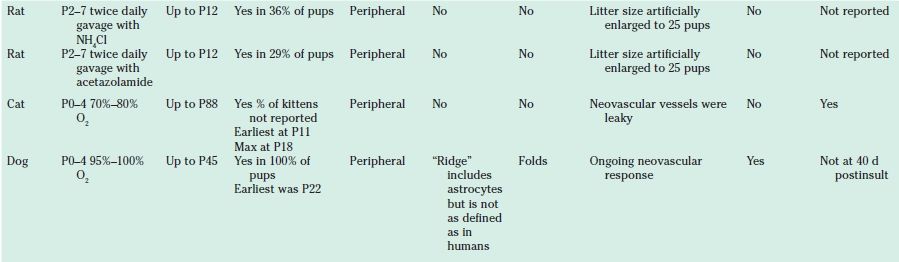
ROP, Retinopathy of prematurity.
whole retinas from animals. The retina is also developing and differentiating, and these processes create oxygen demands within different retinal regions at various times. These caveats are considered when choosing animal models to address hypotheses and when designing experiments and interpreting data.
Retinas can be removed from eyes, fattened, and labeled with stains to visualize the vasculature in retinal fat mounts. Areas of normal retinal vascularization, intravitreal neovascularization, and avascular retina can be measured as compared to total retinal area. There are two broad categories of models: those in which high oxygen damages newly formed capillaries and variable oxygen models in which oxygen fluctuations delay ongoing vasculature. Each is useful to address different experimental questions (Fig. 43.2).

FIGURE 43.2 The features of retinal vessels visualized by retinal flat mount with isolectin staining in models of OIR. In mouse OIR model, mice pups are exposed to 75% O2 from postnatal day 7 to day 12. This model produces vaso-obliteration of newly formed capillaries in the posterior retina at day 12 with decreased retinal vascular endothelial growth factor (VEGF), IGF-1, and erythropoietin (EPO) and intravitreal neovascularization at the junction of vascular and avascular retina at day 17 with increased retinal IGF-1, VEGF, ROS, and EPO signaling. In rat 50/10 OIR model, newborn rat pups are exposed to 50% O2 followed by 10% O2 cycled by 24 hours until postnatal day 14. This model produces features of ROP including delayed retinal vascularization at peripheral retina with decreased retinal EPO and IGF-1 and intravitreal neovascularization at the junction of vascular and avascular retina with increased VEGF, IGF-1, and ROS signaling.
Hyperoxia-Induced Vaso-Obliteration Followed by Relative Hypoxia
Initial OIR models tested the role of high oxygen at birth on the development of ROP (Table 43.1). The mouse OIR model developed by Smith and D’Amore is the most recognized and used (10). Mice are exposed to constant oxygen (75%) at day 7 of life. High oxygen can change the fate of endothelial precursors (26) and cause newly developed capillaries to constrict in part, from apoptotic death of endothelial cells, which leads to avascular retina. In these animal models, this event is termed vaso-obliteration and occurs in the central, previously vascularized retina (10). Following return to room air, endothelial budding into the vitreous is driven by relative hypoxia in the central avascular retinal regions (22). The mouse OIR model is particularly valuable to study genetic mechanisms and the effects of high oxygen stresses and relative retinal hypoxia on angiogenesis, potentially as what occurred in ROP of the 1940s. However, today, high oxygen at birth is avoided in neonatal units that have implemented technology to monitor and regulate oxygen. High oxygen remains a concern, for example, in units in which resources are insufficient to implement oxygen monitoring and regulation (27).
Variable Oxygen
Variable oxygen is common in neonatal units that regulate oxygen. There are a number of models of variable oxygen stresses (28–30) (Table 43.1). The most characterized is the rat 50/10 OIR model developed by John Penn, in which newborn rat pups are exposed to 50% oxygen for 24 hours, followed by 10% oxygen for 24 hours (13). The 24-hour cycles are repeated until day 14, at which time animals are returned to room air. At postnatal day 14, there is peripheral avascular retina and by day 18, aberrant intravitreal neovascularization at the junction of vascular and avascular retina. The rat 50/10 OIR model may simulate the pattern of oxygen exposure experienced by preterm infants in the United States today. The oxygen extremes cause arterial oxygen levels similar to transcutaneous oxygen levels measured in human preterm infants (5). The appearance of the retina is similar to zone II, stage 3 ROP with a central vascularized retina and a peripheral avascular area. Hypoxic retina is present in the avascular peripheral retina as well as in the vascularized regions around retinal vessels (20).
Signaling Pathways Activated by Postnatal Stresses in Animal Models
Oxidative Signaling Pathways
Using OIR models, it has been found that retinal ROS are generated in association with avascular retinal areas and pathologic disordered angiogenesis leading to intravitreal neovascularization (stage 3 ROP) (20,31). Many signaling pathways can be triggered by oxidative compounds and lead to apoptosis, angiogenesis, and inflammation (5). Intravitreal lipid hydroperoxides (LHP), end products of ROS, induced a cascade of angiogenic growth factors and cytokines in the rabbit eye and led to the development of intravitreal neovascularization (32). Using the 50/10 OIR model, a trend toward increased LHP was found in temporal association with the development of intravitreal neovascularization (33). However, neither intravitreal neovascularization nor avascular retina was reduced by the broad antioxidant, N-acetylcysteine (NAC), at a dose that inhibited LHP production in the retina (33). However, in a mouse model of OIR, NAC given during hyperoxia reduced both vaso-obliteration and later intravitreal neovascularization (32).
Treatment with vitamin C or E (5,34) or liposomal superoxide dismutase (35) reduced peripheral avascular retina and vascular leakage and increased capillary density compared to sham-injected controls but did not reduce pathologic intravitreal neovascularization in the 50/10 OIR model.
Several studies have looked at the effect of the NADPH oxidase inhibitor, apocynin, on both avascular retina and intravitreal neovascularization. In both the mouse OIR and rat 50/10 OIR models, apocynin reduced avascular retina and apoptosis (33,36). Using the mouse model of OIR, activated NADPH oxidase induced VEGF expression and intravitreal neovascularization (34), in part, by inhibiting the anti-inflammatory effect of the transcription factor, peroxisome proliferator–activated receptor gamma (37). In the 50/10 OIR model rescued with supplemental oxygen instead of room air, activated NADPH oxidase mediated intravitreal neovascularization (20), in part, through Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling (38).
Besides NADPH oxidase, other enzymes catalyze ROS generation in endothelial cells and include eNOS and cyclooxygenase. eNOS catalyzes the release of nitric oxide (NO). NO is an important vasodilator and has both protective and proangiogenic properties in the eye that preserve endothelial cell barrier integrity by reducing apoptosis. However, NO can react with ROS and cause retinal microvascular degeneration by processing nitro-oxidative stress and generating damaging peroxynitrite (39,40). Retinal peroxynitrite was significantly elevated in the mouse model of OIR and caused capillary endothelial cell apoptosis leading to retinal vessel loss during hyperoxia. Exogenous peroxynitrite caused angiogenesis in vitro and in vivo by enhancing VEGF signaling during relative hypoxia in the mouse OIR (31). Enos-/- mice subjected to the OIR model developed less avascular retina and intravitreal neovascularization, providing evidence of the importance of eNOS and nitro-oxidative stress in ROP (39). However, there was no change in retinal VEGF levels in Enos-/- mice suggesting that eNOS-regulated nitro-oxidative stress did not mediate VEGF expression in this model (39). In the 50/10 OIR model, activated eNOS was found in association with increased arteriolar tortuosity. However, eNOS was not found to be activated by VEGF (37,38). Reactive nitrogen species, such as NO2*, can isomerize arachidonic acid to trans–arachidonic acid, which contributes to hyperoxia-induced vaso-obliteration by upregulating the antiangiogenic agent, thrombospondin-1, in the mouse model of OIR. Retinal polyunsaturated fatty acids, prevalent throughout particularly in photoreceptor outer segments, are susceptible to trans–arachidonic acid formation when arachidonic acid interacts with reactive nitrogen species (41). Therefore, evidence suggests that ROS signaling can cause pathologic features independent of and in association with VEGF signaling.
Bringing It Back to the Infant: Clinical Studies of Antioxidants on ROP: The signaling of oxidative compounds is evolving and complex, but given the preterm infant’s predisposition to increased ROS and the evidence from animal models of oxygen stresses, it appears that oxidative stress does contribute to features of severe ROP. Several clinical studies were done to test the effect of vitamin E supplementation on ROP in infants, but many of the studies were inconclusive or stopped because of complications, including neonatal sepsis and necrotizing enterocolitis (42). A meta-analysis of these studies showed that vitamin E supplementation reduced the risk of the stage 3 ROP by 52% (43). A clinical trial testing low-dose NAC (16 to 32 mg/kg/d) in premature infants born under 1,000 g birth weight for the first 6 days after birth showed no significant difference in survival, body weight gain, respiratory disease, or ROP (24). A randomized and double-blind clinical study tested the effect of lutein administered to preterm infants born 29.9 ± 1.9 weeks’ gestational age and 1,331 ± 415 g birth weight but found no effect on the incidence of ROP at any stage compared to control (44).
Vascular Endothelial Growth Factor and Other Angiogenic Factors
A number of angiogenic factors are involved in models of OIR and include VEGFA, EPO, IGF-1, angiopoietins, adrenomedullin, stromal-derived factor, and hypoxia inducible factor-1α (HIF-1α). Of these, VEGFA is perhaps the best studied in the pathogenesis of ROP. VEGFA was identified originally as a vasopermeability factor and later as an angiogenic factor (45). The human VEGFA gene produces six splice variants through alternative mRNA splicing (46). Due to differences in the affinity for heparin binding, the splice variants were found to have different biologic functions (47,48). VEGF165 is a predominant splice variant and creates a chemoattractant gradient for migrating endothelial cells during retinal vascular development (49), but also causes inflammatory leukostasis, endothelial apoptosis, and avascular retina, as well as pathologic angiogenesis (50). VEGF promotes angiogenesis by signaling through its receptors (VEGFRs). There are three known VEGFRs and several coreceptors, which are VEGFR1 (Flt-1), VEGFR2 (KDR/Flk-1), VEGFR3 (Flt4), and the neuropilins (NRP1 and NRP2). VEGFR1 has very high affinity for VEGF binding. VEGFR2 has high affinity for VEGF binding, but less than VEGFR1, and VEGFR3 only binds VEGFC and VEGFD. All the VEGFRs are members of the ft subfamily of receptor tyrosine kinases. VEGFR2 has strong tyrosine kinase activity and is the receptor believed to be most involved with angiogenic processes (51). VEGFRs are located on endothelial cells, but in the early developing retina of mouse, VEGFR1 and VEGFR2 are also localized in retinal neurons of the inner nuclear and ganglion layers. NRPs act as coreceptors for specific VEGFRs. NRP1 is a coreceptor for VEGFR2 and enhances the binding and biologic activity of VEGF165.
An increase in VEGFA protein was found in the ocular fluid of human infants with ROP (48), and VEGF mRNA was detected in the avascular retina of a human infant with stage 3 ROP (49). Inhibition of VEGF bio-activity with a monoclonal antibody against VEGF reduced severe ROP (52). However, VEGF is also essential in retinal vascular development and as an endothelial and neuronal survival factor (53,54). VEGF signaling regulates the number and length of filopodia in endothelial tip cells (55). The filopodia are considered the guidance-seeking protrusions from neurons and may serve a similar role in endothelial tip cells at the migrating front during retinal vascular development. VEGF signaling also interacts with the delta-like ligand 4/ Notch1 signaling pathway, regulating the endothelial tip-to-stalk cell ratio at the junction of the vascular and avascular retina. This ratio is important in ordered developmental angiogenesis (56), and proliferation of stalk cells can cause intravitreal neovascularization (57). An axon guidance signaling Slit2/Roundabout 4 (Robo4) pathway has been found important in stabilizing retinal vessels by inhibiting VEGF-induced pathologic angiogenesis (58). Evidence also supports that the receptors VEGFR1 and VEGFR2 are required for normal neural retinal development (59).
VEGF is upregulated by a number of stimuli that affect preterm infants, including hypoxia, ROS, and inflammatory cytokines. Hypoxia-induced VEGF expression is regulated by mechanisms including transcriptional activation, alternative translational initiation, and mRNA stabilization. The transcriptional activation involves stabilization of the subunit HIF-α. In normoxia, HIF-α is hydroxylated by prolyl hydroxylase enzymes (60). However, hypoxic conditions lower the activity of prolyl hydroxylases, preventing HIF-α from hydroxylation. The stabilized HIF-α translocates to nucleus and heterodimerizes with HIF-β subunit acting as a transcriptional factor to regulate gene transcription. In the mouse model of OIR, inhibition of prolyl hydroxylases promoted stabilization of HIF during hyperoxia. This then caused less vaso-obliteration, less endothelial budding into the vitreous, and reduced vessel tortuosity in the model (61). VEGF Signaling: In the mouse model of OIR, high constant inspired oxygen at 75% led to reduced retinal VEGF expression in association with vaso-obliteration (Fig. 43.2). Prior to this phase, if VEGF (62), placental growth factor (63–65), IGF-1 (65), IGF-1–binding protein 3 (IGFBP3) (66), EPO (67), omega-3 fatty acids (68), and certain peptides (69) were given to mice, vaso-obliteration was reduced. When mouse pups were then placed into room air following high oxygen, relative tissue hypoxia occurred in the vaso-obliterated retina, followed by overexpression of VEGF (70) and EPO (71) and endothelial budding above the internal limiting membrane.
In the rat 50/10 OIR model, repeated fluctuations in oxygen cycled between 50% and 10% increased retinal VEGF164 (rat homolog to human VEGF165) expression at both mRNA and protein levels beginning at day 8 of life compared to that in the pups raised in room air or to a single hypoxic episode (10% O2) (72). Retinal VEGF was increased in association with persistent avascular retina, suggesting that increased VEGF was not able to support ongoing physiologic retinal vascularization. When rat pups were returned to room air from oxygen cycling, retinal VEGF was increased compared to the same aged pup raised in room air, in association with intravitreal neovascularization (73).
Retinal hypoxia has been measured in the rat OIR model following repeated fluctuations in oxygen and in the mouse OIR model when mice are brought out of hyperoxia into room air (20,21). Retinal hypoxia can cause VEGF-induced aberrant intravitreal neovascularization, and treatment with agents that block the bioactivity of VEGF can reduce tortuosity of vessels (analogous to plus disease) (38) and later intravitreal neovascularization (37,38,64,70,74–78).
In the mouse OIR, inhibition of VEGF reduced endothelial budding into the vitreous during the vasoproliferative phase and did not appear to affect the avascular retina; however, the area of avascular retina was not quantified (79). In the rat 50/10 OIR model, inhibition of retinal VEGF by a neutralizing antibody against VEGF164 significantly reduced clock hours of intravitreal neo-vascularization by 50% and did not interfere with retinal vascular development (37). At first the finding that an antiangiogenic compound would not interfere with ongoing vascular development at certain doses seemed counterintuitive. However, further study showed that VEGF neutralization affected the orientation of dividing vascular cells. A single allele knockout of a splice variant or receptor in the VEGF signaling pathway is lethal. Therefore, to study the effect of excessive VEGF signaling on endothelial cell divisions, investigators used an embryonic stem cell model that had a Flt-1 knockout (knockout to VEGFR1). VEGFR1 acts as a decoy receptor in development, limiting the amount of VEGF available to signal through the angiogenic, VEGFR2. In the Flt-1 knockout model, VEGF could not bind VEGFR1 and triggered signaling through VEGFR2. The orientation of the cleavage planes of dividing endothelial cells to the long axes of vessels predicts the future location of daughter cells (Fig. 43.3). If cells divided and the cleavage planes were parallel to the long axis of the vessels, then the resulting vessels were dilated. If the cleavage planes of the dividing endothelial cells were perpendicular to the long axis of the vessels, vessels were elongated. When cleavage planes were disordered relative to the long axis of the vessels, the result was disordering of the vasculature. Ordered angiogenesis was restored in the Flt-1-/- when a CD31 transgene containing Flt-1 was introduced to the model, thus restoring the ability to bind excess VEGF and limit angiogenic signaling (80). In an analogous study in the 50/10 OIR model, the angles between cleavage planes of dividing vascular endothelial cells and the long axes of the vessels (i.e., cleavage angles) were found to be less likely to predict lengthening of the retinal vessels. Vessels were also more tortuous in the 50/10 OIR model than those in room air. A neutralizing antibody to VEGF, given at a dose found to reduce intravitreal neovascularization, restored cleavage angles to predict lengthening of vessels and reduced tortuosity (38). This study suggests a way in which excessive VEGF signaling may lead to disordering of dividing endothelial cells and that restoration of a normal level of signaling may facilitate ordered retinal vascular development and reduce peripheral avascular retina. However, dose appears important (78). In the beagle model, a low dose of a VEGFtrap reduced intravitreal neovascularization without adversely affecting ongoing retinal vascular development; however, a high dose caused persistent avascular retina (78).
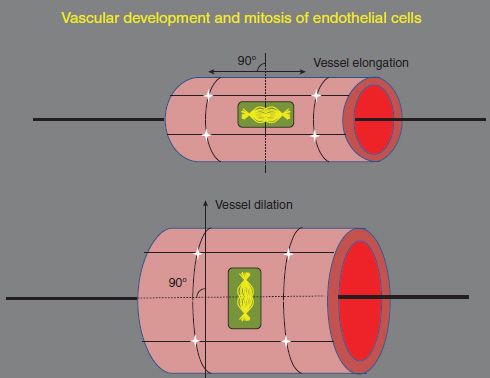
FIGURE 43.3 Diagram of the orientation of the cleavage planes of dividing endothelial cells and vascular development. Vessel elongation results when the cleavage planes of the dividing endothelial cells are perpendicular to the long axis of the vessels, and when the cleavage planes of dividing endothelial cells are parallel to the long axis of the vessels, the vessels are elongated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


